How to read this manual
Too lazy or too busy to read the manual? Click here for an LLM-friendly version of it. You can simply download this raw manual, upload it into your LLM of choice and ask for guidance if you get stuck. Do not trust it blindly though, LLMs often miss important things!
This is a manual for the gorder tool—a command-line (CLI) application, Python package, and Rust crate for calculating lipid order parameters from molecular simulations. Most of the manual focuses on the CLI application.
The contents of this manual are shown in the panel on the left side of the page.
In the Introduction section, we discuss why gorder exists and what lipid order parameters even are. Feel free to skip this section.
In the Beginner section, we describe how to install gorder and run basic analyses of atomistic, coarse-grained, and united-atom systems. If you are only interested in, for example, atomistic systems, you can skip the pages dedicated to coarse-grained and united-atom analyses.
In the Advanced section, we discuss various features of gorder, such as calculating order parameters for individual leaflets, constructing order parameter maps, estimating analysis errors, calculating order parameters in a specific part of the membrane, and analyzing simulations using dynamically estimated membrane normals. The individual pages are quite independent, so you can skip directly to the ones you need.
In the Expert section, we cover more advanced features of gorder, which mostly provide greater control over the analyses. This includes manual lipid assignment to leaflets, manual specification of membrane normals, and ignoring periodic boundary conditions. You will most likely not need these features.
In the APIs section, we briefly explain how to use gorder directly as a Python package or a Rust crate. These topics also have their own dedicated manuals (Python, Rust). The CLI version of gorder is fully featured; you do not need to use the APIs unless you want direct integration with your code.
In the Meta section, we discuss the limitations of gorder (you should read this page!), where to send your feedback, and how to cite gorder if you use it in a scientific project.
What is gorder?
gorder is a command-line tool that aims to provide a comprehensive, reliable, simple, and fast way to calculate atomistic, coarse-grained, and united-atom lipid order parameters from Gromacs simulations.
The primary (and possibly naïve) goal of gorder is to completely replace the use of gmx order. gmx order is a tool still widely used in the Gromacs community despite its tedious usage and the fact that it sometimes produces completely incorrect results.1
Apart from calculating the order parameters correctly, gorder also offers many additional features, such as reporting order parameters for individual C-H bonds, error estimation, leaflet classification, geometric selection, or construction of ordermaps. gorder can also calculate order parameters for non-planar membranes such as tubes or vesicles.
gorderis written in Rust and is also available as a Python package and a Rust crate. If you want to see the code (or comparison with other programs), visit thegorderGitHub repository.
Even Gromacs developers say that you should not use gmx order.
Some theory
Lipid order parameters quantify the degree of ordering in lipid membranes. They can be obtained from both experimental measurements and simulations, making them quite useful for checking force-field accuracy, studying molecular structures, tracking phase transitions and various other stuff.
The order parameter in atomistic systems can be calculated as:
where is the angle between the C-H bond vector and the bilayer normal, and the angular brackets represent molecular and temporal ensemble averages. The order parameter for atomistic systems is usually reported as the negative of this calculated value, (often denoted as or ). Higher positive values of indicate more ordered membrane structures.
In coarse-grained systems, the order parameter can be calculated using the same equation. However, here corresponds to the angle between the vector connecting two consecutive beads of the lipid and the bilayer normal. Using this definition, the order parameter (sometimes denoted as ) increases as the membrane structure becomes more ordered. For coarse-grained systems, the order parameter is usually reported directly as to facilitate comparison with the atomistic (which follows the same trend).
In united-atom systems, the situation is more complex because we aim to calculate order parameters for C-H bonds, but the positions of hydrogen atoms are not a priori known. To determine the order parameters, the positions of hydrogen atoms can either be explicitly predicted or implicitly derived during the calculation of lipid order parameters (see Piggot et al. for more details). gorder employs the former approach, explicitly predicting the positions of hydrogen atoms using the same method as the NMR Lipids-associated buildH tool. Once the hydrogen positions are predicted, the order parameters are calculated in the same manner as for atomistic systems.
Installation
Installing the gorder tool consists of the following steps:
-
Install Rust.
- Follow this guide for your operating system.
- If you already have Rust installed, you can skip this step. To check, run
rustc --version.
You need Rust v1.82 or newer to be able to install
gorder. To update Rust, runrustup update. -
Install
gorder.cargo install gorder
You can verify that the installation was successful by running gorder --version. The current version of the gorder tool should be displayed in the terminal. If everything went right, you can now call gorder from anywhere.
Troubleshooting
Below are some common errors you might encounter when installing gorder on Linux. If you are still unable to proceed, please open an issue on GitHub and provide details about the problem.
Unfortunately, we cannot provide support for installing
gorderon Windows or macOS, and we apologize for any inconvenience this may cause.
Command not found: cargo
If, after installing Rust, you are unable to run cargo install gorder and receive an error like command not found: cargo, it likely means that cargo is not included in your PATH. Try closing and reopening your terminal.
If this does not help and you are using bash, try adding the following line to your .bashrc file (typically located in your home directory, ~): export PATH="$HOME/.cargo/bin:$PATH". Then restart the terminal again.
Atomistic order parameters
Preparing the input
Suppose we have a CHARMM36 membrane composed of POPC, POPE, and POPG lipids.
To calculate atomistic order parameters, we need two Gromacs files:
- A TPR file containing the system structure and topology (
system.tpr). - An XTC trajectory file (
md.xtc) whose frames will be analyzed.
It is recommended to use TPR and XTC files, but
gorderalso supports some other file formats.
Next, we create a configuration YAML file that specifies the options for the analysis:
structure: system.tpr
trajectory: md.xtc
analysis_type: !AAOrder
heavy_atoms: "@membrane and name r'C3.+|C2.+'"
hydrogens: "@membrane and element name hydrogen"
output: order.yaml
In the configuration YAML file, the analysis type AAOrder requires you to specify both heavy_atoms and hydrogens. gorder will then identify all bonds connecting the selected heavy atoms with the selected hydrogen atoms. The order parameters are calculated for all these identified bonds.
The atoms are selected using a query language called GSL. If you are familiar with the query language used in VMD, you'll find the basic syntax of GSL intuitive.
Here:
heavy_atomsare selected using the query@membrane and name r'C3.+|C2.+', which selects all palmitoyl and oleoyl carbons of the membrane lipids.hydrogensare selected using the query@membrane and element name hydrogen.
@membraneis a GSL autodetection macro that selects all atoms of common membrane lipids.r'C3.+|C2.+'is a regular expression block, natively supported by GSL.
The results of the analysis will be saved in the order.yaml file as (see Theory).
Running the analysis
We save the configuration YAML file, for example, as analyze.yaml. Then, we run gorder as follows:
$ gorder analyze.yaml
During the analysis, we will see something like this:

Note that the structure from the TPR file is not analyzed. The TPR file is only used to construct the system and obtain its topology.
The results of the analysis are saved in the order.yaml file. Here is an excerpt from the file:
# Order parameters calculated with 'gorder v1.2.0' using a structure file 'system.tpr' and a trajectory file 'md.xtc'.
average order:
total: 0.1631
POPE:
average order:
total: 0.1601
order parameters:
POPE C22 (23):
total: 0.1036
bonds:
POPE H2R (24):
total: 0.0876
POPE H2S (25):
total: 0.1196
POPE C32 (32):
total: 0.2297
bonds:
POPE H2X (33):
total: 0.2423
POPE H2Y (34):
total: 0.2171
# (...)
POPC:
average order:
total: 0.166
order parameters:
POPC C22 (32):
total: 0.1109
bonds:
POPC H2R (33):
total: 0.0935
POPC H2S (34):
total: 0.1283
POPC C32 (41):
total: 0.2373
bonds:
POPC H2X (42):
total: 0.2483
POPC H2Y (43):
total: 0.2264
# (...)
POPG:
average order:
total: 0.1608
order parameters:
POPG C22 (25):
total: 0.0987
bonds:
POPG H2R (26):
total: 0.08
POPG H2S (27):
total: 0.1174
POPG C32 (34):
total: 0.2272
bonds:
POPG H2X (35):
total: 0.2367
POPG H2Y (36):
total: 0.2177
gorder automatically identified three molecule types and all relevant bonds. Order parameters are reported separately for each molecule type: for each bond type of each molecule type and for each heavy atom type of each molecule type. Order parameters for heavy atom types are obtained by averaging the order parameters of their bonds with hydrogens. average order corresponds to the average order of all the relevant bonds of the entire system or a single molecule type, respectively.
⚠️ The atom types are listed in the same order as they appear in the input TPR structure. Note that in some force fields (e.g., CHARMM), the sequence of atoms may be unintuitive (e.g., C32 can appear between C22 and C23, even though they are from different lipid tails). Always check the output before plotting the results!
Let's take a closer look at a part of the output YAML file:
average order:
total: 0.1631 # average order calculated for all molecules in the entire membrane
POPE: # name of the molecule type
average_order:
total: 0.1601 # average order calculated for POPE molecules in the entire membrane
order parameters:
POPC C22 (23): # heavy atom type: residue atom_name (relative_index)
total: 0.1036 # order parameter of the heavy atom
bonds: # bonds of this heavy atom with hydrogens
POPC H2R (24): # hydrogen type: residue atom_name (relative_index)
total: 0.0876 # order parameter of bond with this hydrogen
POPC H2S (25): # hydrogen type: residue atom_name (relative_index)
total: 0.1196 # order parameter of bond with this hydrogen
YAML files are easy to read programmatically and not completely human-unreadable. However, gorder also provides other output formats (XVG, CSV, human-readable table). See Output formats for more information.
Using groups from an NDX file
gorder also supports using groups from NDX files. Let's suppose our NDX file already contains a group called TailCarbons, which specifies all the carbons to use. We no longer need to specify them using the complex regular expression from the previous example and can simply select the group. Our configuration YAML file will look like this:
structure: system.tpr
trajectory: md.xtc
index: index.ndx # name of the NDX file
analysis_type: !AAOrder
heavy_atoms: "TailCarbons" # name of a group from NDX file
hydrogens: "@membrane and element name hydrogen"
output: order.yaml
Then, we run the analysis in the same way as before.
Coarse-grained order parameters
Preparing the input
Suppose we have a Martini 3 membrane composed of POPC, POPE, and POPG lipids.
To calculate coarse-grained order parameters, we need two Gromacs files:
- A TPR file containing the system structure and topology (
system.tpr). - An XTC trajectory file (
md.xtc) whose frames will be analyzed.
It is recommended to use TPR and XTC files, but
gorderalso supports some other file formats.
Next, we create a configuration YAML file that specifies the options for the analysis:
structure: system.tpr
trajectory: md.xtc
analysis_type: !CGOrder
beads: "@membrane"
output: order.yaml
In the configuration YAML file, the analysis type CGOrder requires you to specify beads that will be considered for the analysis. gorder will then identify all bonds connecting the selected beads. The order parameters are calculated for all these identified bonds.
The atoms are selected using a query language called GSL. If you are familiar with the query language used in VMD, you'll find the basic syntax of GSL intuitive.
Here beads are selected using the query @membrane. That is a GSL autodetection macro that select all beads or atoms of common membrane lipids.
The results of the analysis will be saved in the order.yaml file as (see Theory).
Running the analysis
We save the configuration YAML file, for example, as analyze.yaml. Then, we run gorder as follows:
$ gorder analyze.yaml
During the analysis, we will see something like this:

Note that the structure from the TPR file is not analyzed. The TPR file is only used to construct the system and obtain its topology.
The results of the analysis are saved in the order.yaml file. Here is an excerpt from the file:
# Order parameters calculated with 'gorder v1.2.0' using a structure file 'system.tpr' and a trajectory file 'md.xtc'.
average order:
total: 0.2937
POPC:
average order:
total: 0.2902
order parameters:
POPC NC3 (0) - POPC PO4 (1):
total: -0.1362
POPC PO4 (1) - POPC GL1 (2):
total: 0.585
POPC GL1 (2) - POPC GL2 (3):
total: -0.1808
POPC GL1 (2) - POPC C1A (4):
total: 0.3835
# (...)
POPE:
average order:
total: 0.2959
order parameters:
POPE NH3 (0) - POPE PO4 (1):
total: -0.1293
POPE PO4 (1) - POPE GL1 (2):
total: 0.6072
POPE GL1 (2) - POPE GL2 (3):
total: -0.1833
POPE GL1 (2) - POPE C1A (4):
total: 0.3965
# (...)
POPG:
average order:
total: 0.3063
order parameters:
POPG GL0 (0) - POPG PO4 (1):
total: 0.0004
POPG PO4 (1) - POPG GL1 (2):
total: 0.5917
POPG GL1 (2) - POPG GL2 (3):
total: -0.1807
POPG GL1 (2) - POPG C1A (4):
total: 0.395
# (...)
gorder automatically identified three molecule types and all relevant bonds. Order parameters are reported for each bond type of each molecule type. average order corresponds to the average order of all the relevant bonds of the entire system or a single molecule type, respectively.
⚠️ The bond types are listed in the same order their atoms appear in the input TPR structure. Note that in some force fields, the sequence of atoms may be unintuitive and bonds from different tails may not be separated. Always check the output before plotting the results!
Let's take a closer look at a part of the output YAML file:
average order:
total: 0.2937 # average order calculated for all molecules in the entire membrane
POPC: # name of the molecule type
average order:
total: 0.2902 # average order calculated for POPC molecules in the entire membrane
order parameters:
POPC NC3 (0) - POPC PO4 (1): # bond type (atom type 1 - atom type 2)
total: -0.1362 # order parameter of this bond
POPC PO4 (1) - POPC GL1 (2): # atom types are specified as residue atom_name (relative_index)
total: 0.585
POPC GL1 (2) - POPC GL2 (3):
total: -0.1808
POPC GL1 (2) - POPC C1A (4):
total: 0.3835
YAML files are easy to read programmatically and not completely human-unreadable. However, gorder also provides other output formats (XVG, CSV, human-readable table). See Output formats for more information.
Using groups from an NDX file
gorder also supports using groups from NDX files. Let's suppose our NDX file already contains a group called LipidBeads, which specifies all the beads to use. We can then simply select the group. Our configuration YAML file will look like this:
structure: system.tpr
trajectory: md.xtc
index: index.ndx # name of the NDX file
analysis_type: !CGOrder
beads: "LipidBeads" # name of a group from NDX file
output: order.yaml
Then, we run the analysis in the same way as before.
United-atom order parameters
Preparing the input
Suppose we have a membrane composed of POPC Berger lipids.
To calculate united-atom order parameters, we need two Gromacs files:
- A TPR file containing the system structure and topology (
system.tpr). - An XTC trajectory file (
md.xtc) whose frames will be analyzed.
It is recommended to use TPR and XTC files, but
gorderalso supports some other file formats.
Next, we create a configuration YAML file that specifies the options for the analysis:
structure: system.tpr
trajectory: md.xtc
analysis_type: !UAOrder
# all carbons of the lipid except for carboxyl atoms (C15, C34) and unsatured atoms (C24, C25)
saturated: "@membrane and element name carbon and not name C15 C34 C24 C25"
unsaturated: "@membrane and name C24 C25"
output: order.yaml
In the configuration YAML file, you must specify which carbons should have their order parameters calculated and whether they are saturated or unsaturated. gorder will automatically construct hydrogens for saturated carbons so that the total number of bonds of each carbon is 4. For example:
- Carbons within the acyl chains (bonded to two other carbons) will be assigned two hydrogens, whose positions are predicted based on the lipid's geometry.
- Methyl carbons at the ends of the acyl chains will have three hydrogens constructed.
- The central carbon of the glycerol will have one hydrogen constructed.
Unsaturated carbons are those with a double bond to another carbon in the acyl chain (and one single bond to another carbon). Only one hydrogen is predicted for such atoms.
In configuration YAML file above, we calculate order parameters for all carbons in POPC lipids (except for the carboxyl atoms C15 and C34, which lack hydrogens and should be explicitly excluded, as explained below). We also specify a double bond between atoms C24 and C25.
Positions of hydrogens are predicted in exactly the same way as in the
buildHtool. See the buildH documentation for more information.
The results of the analysis will be saved in the order.yaml file as (see Theory).
Limitations to consider
There are some limitations you should be aware of when calculating united-atom lipid order parameters:
- Order parameters calculated for individual C-H bonds of methyl carbons are not reliable. Please read the note on order parameters of methyl groups.
- While the calculation of atomistic and coarse-grained order parameters can be performed for all atom types, the calculation of united-atom parameters must be exclusively performed for carbon atoms.
gorderdoes not have access to the multiplicities of covalent bonds (i.e., it cannot automatically distinguish between single and double bonds). This is why you must specify saturated and unsaturated carbons separately, and why you should exclude carboxyl carbons from the analysis. A carboxyl carbon has a double bond to oxygen and is not bonded to a hydrogen. However, sincegordercannot detect the multiplicity of the double bond, it will attempt to add a hydrogen to the carbon if it is included among saturated carbons. Therefore, you should either exclude the carboxyl carbon entirely or include it among unsaturated atoms.- If you are calculating united-atom order parameters for unnatural or fictional molecules,
gordermay not work properly.gorderonly supports standard lipid molecules, where:- No carbon atom is bonded to two double bonds simultaneously.
- No triple bonds are present.
- Terminal carbons are methyl groups, not methylidenes.
- Carbon-hydrogen bonds have a length of about 0.109 nm.
Running the analysis
We save the configuration YAML file, for example, as analyze.yaml. Then, we run gorder as follows:
$ gorder analyze.yaml
During the analysis, we will see something like this:

Note that the structure from the TPR file is not analyzed. The TPR file is only used to construct the system and obtain its topology.
The results of the analysis are saved in the order.yaml file. Here is an excerpt from the file:
# Order parameters calculated with 'gorder v1.2.0' using a structure file 'system.tpr' and a trajectory file 'md.xtc'.
average order:
total: 0.098
POPC:
average order:
total: 0.098
order parameters:
POPC C1 (0):
total: 0.0001
bonds:
- total: 0.0001
- total: -0.002
- total: 0.0021
POPC C2 (1):
total: 0.0001
bonds:
- total: -0.0
- total: 0.0022
- total: -0.0019
POPC C3 (2):
total: 0.0001
bonds:
- total: -0.0001
- total: -0.0021
- total: 0.0025
POPC C5 (4):
total: -0.0554
bonds:
- total: -0.0665
- total: -0.0443
# (...)
POPC C24 (23):
total: 0.0723
bonds:
- total: 0.0723
POPC C25 (24):
total: 0.0046
bonds:
- total: 0.0046
# (...)
POPC CA2 (51):
total: 0.0254
bonds:
- total: -0.0381
- total: 0.0572
- total: 0.0572
gorder automatically identified one molecule type and all relevant bonds. It then predicted the positions of missing hydrogens for the individual carbons. Order parameters are reported for each carbon of each molecule type, as well as for each predicted C-H bond. average_order corresponds to the average order of all the relevant atoms of the entire system or a single molecule type, respectively.
⚠️ The atom types are listed in the same order as they appear in the input TPR structure. Note that in some force fields, the sequence of atoms may be unintuitive and atoms from different tails may not be separated. Always check the output before plotting the results!
Let's take a closer look at a part of the output YAML file:
average order:
total: 0.098 # average order calculated for all molecules in the entire membrane
POPC: # name of the molecule type
average order:
total: 0.098 # average order calculated for POPC molecules in the entire membrane
order parameters:
POPC C1 (0): # carbon type: residue atom_name (relative_index)
total: 0.0001 # order parameter of the carbon
bonds: # bonds of this heavy atom with predicted hydrogens
- total: 0.0001 # order parameter of C-H bond number 1 (predicted hydrogens have no names)
- total: -0.002 # order parameter of C-H bond number 2
- total: 0.0021 # order parameter of C-H bond number 3
YAML files are easy to read programmatically and not completely human-unreadable. However, gorder also provides other output formats (XVG, CSV, human-readable table). See Output formats for more information.
Note on order parameters of methyl groups
This is important! Please read at least the information in this box. You should not trust the order parameters reported for individual C-H bonds in methyl groups. In methyl groups, only the order parameter calculated for the entire carbon is reliable.
While gorder reports order parameters for individual predicted C-H bonds, you might notice that the order parameters for bonds of methyl carbons are somewhat suspicious. Typically, the individual C-H bonds of the same atom have very similar values, but this is not the case here, where the order parameters might differ significantly. For example:
POPC CA2 (51):
total: 0.0254
bonds:
- total: -0.0381 # suspicious
- total: 0.0572
- total: 0.0572
This discrepancy is an artifact of the hydrogen-building process. The results for individual C-H bonds of methyl carbons depend on the order of atoms that are used as "helpers". For gorder, this means that changing the order of atoms in your topology may yield completely different results for bonds in methyl groups, even when analyzing the same (but reordered) trajectory.
However, only the order parameters for individual bonds are affected by this artifact. When averaging the order parameters of the individual bonds, the result remains consistent regardless of the helper atom definitions. Thus, the overall order parameter for the atom (the total field in the output YAML file) is reasonable and correct (to the degree united-atom force fields are accurate) and can be safely reported.
Using groups from an NDX file
gorder also supports using groups from NDX files. Let's suppose our NDX file already contains groups called Satured and Unsaturated, which specify all carbons to analyze. We can then simply select the groups. Our configuration YAML file will look like this:
structure: system.tpr
trajectory: md.xtc
index: index.ndx # name of the NDX file
analysis_type: !UAOrder
saturated: "Saturated" # name of the NDX group containing saturated carbons
unsaturated: "Unsaturated" # name of the NDX group containing unsaturated carbons
output: order.yaml
Then, we run the analysis in the same way as before.
Ignoring some atoms
In some cases, such as when calculating united-atom order parameters from an atomistic system or when working with a hybrid AA-UA system, you may want to ignore certain atoms so that they are not considered when calculating bonds, obtaining the number of hydrogens to construct for each carbon, or determining helper atoms for predicting hydrogen positions. To do this, add an ignore keyword to the analysis_type section of your configuration YAML file:
structure: system.tpr
trajectory: md.xtc
analysis_type: !UAOrder
saturated: "@membrane and name r'C3.+|C2.+' and not name C29 C210 C21 C31"
unsaturated: "@membrane and name C29 C210"
ignore: "element name hydrogen"
output: order.yaml
The above YAML configuration can be used to calculate united-atom lipid order parameters for the tails of atomistic (CHARMM36) POPx lipids (lipids with a palmitoyl and an oleoyl tail). The explicit hydrogen atoms will be ignored, and new hydrogens will be predicted instead.
Selecting atoms using GSL
To specify and select atoms (or coarse-grained beads) in gorder, we use the Groan Selection Language (GSL). Its syntax closely matches that used by VMD and various other analysis tools. GSL additionally supports regular expressions and can reference NDX groups from provided NDX files.
For complete GSL syntax and usage examples, see our GSL guide.
Output formats
The YAML output is always generated by gorder, as it contains all the calculated information. See Atomistic order parameters, Coarse-grained order parameters, or United-atom order parameters for more details about this format. However, gorder can also write output in other formats.
⚠️ Ordering of results in the output
Note that results for atom and bond types are always listed in the same order in which the atoms/bonds appear in the input structure file. In some force fields, this sequence may be unintuitive, with atoms or bonds from different tails mixed together.
To reiterate,
gorderdoes NOT list atoms/bonds based on tail allegiance. Always verify the atom/bond sequence in the output before plotting the results. Be especially careful when using CSV and XVG files, as they may tempt you into plotting them directly without any checks at all.
CSV format
CSV format may be useful if you want to work with the results using spreadsheet software such as LibreOffice Calc or Microsoft Excel, or when working with data analysis libraries like Pandas or Polars.
To write the results of the analysis in CSV format, add output_csv: PATH_TO_OUTPUT to your configuration YAML file.
Here is an excerpt from a CSV file:
molecule,residue,atom,relative index,total,hydrogen #1,hydrogen #2,hydrogen #3
POPE,POPE,C22,23,0.1036,0.0876,0.1196,
POPE,POPE,C32,32,0.2297,0.2423,0.2171,
POPE,POPE,C23,35,0.2138,0.2139,0.2138,
POPE,POPE,C24,38,0.2165,0.2158,0.2172,
POPE,POPE,C25,41,0.2290,0.2305,0.2274,
POPE,POPE,C26,44,0.2081,0.2099,0.2063,
POPE,POPE,C27,47,0.1989,0.2001,0.1977,
POPE,POPE,C28,50,0.1176,0.1163,0.1190,
POPE,POPE,C29,53,0.0703,0.0703,,
POPE,POPE,C210,55,0.0598,0.0598,,
(...)
POPE,POPE,C218,78,0.0321,0.0316,0.0327,0.0319
(...)
POPC,POPC,C22,32,0.1109,0.0935,0.1283,
POPC,POPC,C32,41,0.2373,0.2483,0.2264,
POPC,POPC,C23,44,0.2244,0.2237,0.2252,
POPC,POPC,C24,47,0.2230,0.2223,0.2236,
POPC,POPC,C25,50,0.2355,0.2358,0.2352,
POPC,POPC,C26,53,0.2122,0.2134,0.2109,
POPC,POPC,C27,56,0.2031,0.2035,0.2027,
POPC,POPC,C28,59,0.1196,0.1193,0.1199,
POPC,POPC,C29,62,0.0711,0.0711,,
POPC,POPC,C210,64,0.0610,0.0610,,
(...)
POPC,POPC,C218,87,0.0344,0.0340,0.0351,0.0340
(...)
POPG,POPG,C22,25,0.0987,0.0800,0.1174,
POPG,POPG,C32,34,0.2272,0.2367,0.2177,
POPG,POPG,C23,37,0.2088,0.2075,0.2101,
POPG,POPG,C24,40,0.2117,0.2108,0.2126,
POPG,POPG,C25,43,0.2254,0.2255,0.2253,
POPG,POPG,C26,46,0.2039,0.2006,0.2072,
POPG,POPG,C27,49,0.1933,0.1911,0.1954,
POPG,POPG,C28,52,0.1132,0.1148,0.1117,
POPG,POPG,C29,55,0.0637,0.0637,,
POPG,POPG,C210,57,0.0624,0.0624,,
(...)
POPG,POPG,C218,80,0.0330,0.0296,0.0324,0.0369
Each line corresponds to one heavy atom type (or one bond type in the case of coarse-grained systems). The first column corresponds to the molecule name, the second to the residue name, the third to the atom name, the fourth to the relative index of the atom type, the fifth to the order parameter calculated for this atom type, and the following columns correspond to the order parameters of the bonds the heavy atom type is involved in.
Table format
The Table format presents the results of the analysis in a clear, human-readable format.
To write the results of the analysis in Table format, add output_tab: PATH_TO_OUTPUT to your configuration YAML file.
Here is an excerpt from a Table file:
# Order parameters calculated with 'gorder v1.2.0' using a structure file 'system.tpr' and a trajectory file 'md.xtc'.
Molecule type POPE
TOTAL | H #1 | H #2 | H #3 |
C22 0.1036 | 0.0876 | 0.1196 | |
C32 0.2297 | 0.2423 | 0.2171 | |
C23 0.2138 | 0.2139 | 0.2138 | |
C24 0.2165 | 0.2158 | 0.2172 | |
(...)
AVERAGE 0.1601 |
Molecule type POPC
TOTAL | H #1 | H #2 | H #3 |
C22 0.1109 | 0.0935 | 0.1283 | |
C32 0.2373 | 0.2483 | 0.2264 | |
C23 0.2244 | 0.2237 | 0.2252 | |
C24 0.2230 | 0.2223 | 0.2236 | |
(...)
AVERAGE 0.1660 |
Molecule type POPG
TOTAL | H #1 | H #2 | H #3 |
C22 0.0987 | 0.0800 | 0.1174 | |
C32 0.2272 | 0.2367 | 0.2177 | |
C23 0.2088 | 0.2075 | 0.2101 | |
C24 0.2117 | 0.2108 | 0.2126 | |
(...)
AVERAGE 0.1608 |
All molecule types
TOTAL |
AVERAGE 0.1631 |
Each line corresponds to one heavy atom type (or one bond type in the case of coarse-grained systems). The first column corresponds to the name of the atom type, the second to the order parameter calculated for this atom type, and the following columns correspond to the order parameters of the bond the heavy atom type is involved in. The last line in each molecule type (AVERAGE) corresponds to the average order of all the relevant bonds of a single molecule type. The last segment (All molecule types) shows average order parameter calculated for the entire system (all the identified molecules).
XVG format
XVG format may be useful if you want to quickly visualize the results as plots using xmgrace.
To write the results of the analysis in XVG format, add output_xvg: FILE_PATTERN to your configuration YAML file. One XVG file will be created for each molecule. The name of the molecule will be appended to the end of the file stem. For example, if your file pattern is order.xvg and the membrane contains POPC, POPE, and POPG lipid molecules, three XVG files will be generated: order_POPC.xvg, order_POPE.xvg, and order_POPG.xvg.
Here is an excerpt from an XVG file:
# Order parameters calculated with 'gorder v1.2.0' using a structure file 'system.tpr' and a trajectory file 'md.xtc'.
@ title "Atomistic order parameters for molecule type POPC"
@ xaxis label "Atom"
@ yaxis label "-Sch"
@ s0 legend "Full membrane"
@TYPE xy
# Atom C22:
1 0.1109
# Atom C32:
2 0.2373
# Atom C23:
3 0.2244
# Atom C24:
4 0.2230
# Atom C25:
5 0.2355
# Atom C26:
6 0.2122
# Atom C27:
7 0.2031
# Atom C28:
8 0.1196
(...)
XVG files provide the least complete information. The first column is the index of the analyzed heavy atom type (starting from 1), and the second column is the order parameter of the atom type.
Example of a configuration YAML file with all supported outputs requested
structure: system.tpr
trajectory: md.xtc
analysis_type: !AAOrder
heavy_atoms: "@membrane and name r'C3.+|C2.+'"
hydrogens: "@membrane and element name hydrogen"
output_yaml: order.yaml # for yaml format, you can use either `output`, `output_yaml`, or `output_yml`
output_tab: order.tab
output_xvg: order.xvg
output_csv: order.csv
Order parameters for individual leaflets
gorder can calculate order parameters for the entire membrane as well as for the individual leaflets.
To do this, you need to specify a method for classifying lipids into membrane leaflets. By default, gorder assigns lipids to membrane leaflets independently for each analyzed frame (this can be customized, see Classification frequency), making it suitable even for the analysis of membranes where lipids flip-flop between leaflets.
There are four leaflet classification methods available in gorder: global, local, individual, and clustering. In case you are not satisfied with any of them, you can also assign lipids into leaflets manually.
When calculating order parameters for vesicles or similar highly curved membranes, you should always assign lipids using the
clusteringmethod or manually.
Global method for leaflet classification
Reliable for planar membranes and fast. Recommended for most membranes.
In this method, lipid molecules are assigned to membrane leaflets based on the position of their 'head identifier' relative to the global membrane center of geometry. The 'head identifier' is a single atom representing the head of the lipid. If the 'head identifier' is located "above" the membrane center, the lipid is assigned to the upper leaflet; if it is located "below", it is assigned to the lower leaflet.
To use this method, you must specify the 'head identifier' atoms and all atoms that form the membrane. GSL is used to define these selections.
leaflets: !Global
membrane: "@membrane"
heads: "name P"
Here, we use autodetected membrane atoms to calculate the membrane center and select atoms named 'P' (phosphorus atoms of lipids) as head identifiers. Each analyzed lipid must have exactly one head identifier atom; otherwise, an error will occur.
Local method for leaflet classification
Reliable for planar membranes but slow. Recommended for planar membranes if the
globalandindividualmethods do not work for you.
In this method, lipid molecules are assigned to membrane leaflets based on the position of their 'head identifier' relative to the local membrane center of geometry. The local membrane center is calculated using atoms in a cylinder around the 'head identifier'. If the 'head identifier' is located "above" the local center, the lipid is assigned to the upper leaflet; if "below", it is assigned to the lower leaflet.
For this method, you need to specify a selection of head identifiers, all atoms forming the membrane, and the radius of the cylinder used to define the local membrane.
leaflets: !Local
membrane: "@membrane"
heads: "name P"
radius: 2.5
Autodetected membrane atoms will be used to calculate the membrane center. Only atoms within a cylinder of radius 2.5 nm (with infinite height) centered on the 'head identifier' and oriented along the membrane normal will be used for the local center calculation. The atoms named 'P' (phosphorus atoms of lipids) are used as 'head identifiers'.
Individual method for leaflet classification
Less reliable but very fast. Recommended for very large, undulating planar membranes.
In this method, lipid molecules are assigned to membrane leaflets based on the position of their 'head identifier' relative to their 'tail ends'. 'Tail ends' refer to the last heavy atoms or beads of the lipid tails. Each lipid molecule may have multiple 'tail ends', but only one 'head identifier'. If the 'head identifier' is located "above" the 'tail ends', the lipid is assigned to the upper leaflet; if it is located "below", it is assigned to the lower leaflet.
To use this method, you must specify selections for the 'head identifiers' and the 'tail ends':
leaflets: !Individual
heads: "name P"
methyls: "name C218 C316"
In this example, atoms named 'P' (phosphorus atoms of lipids) are used as head identifiers, and 'C218' or 'C316' atoms (the last carbons of oleoyl and palmitoyl chains) are used as tail ends.
Clustering method for leaflet classification
Reliable but very slow. Recommended for curved membranes.
This method assigns lipid molecules to membrane leaflets by clustering their head groups using spectral clustering. This method can handle any membrane geometry that is physically realistic, including curved membranes with pores or lipid flip-flop.
To use the clustering method, specify a selection for 'head identifiers':
leaflets: !Clustering
heads: "name P"
In this example, phosphorus atoms ('P') serve as head identifiers. As with other methods, each lipid molecule must have exactly one head identifier, but you can also include head identifiers for lipid molecules for which you are not calculating the order parameters. The method groups the specified atoms into two clusters representing the membrane leaflets.
Important considerations for the clustering method
- Upper vs lower leaflet assignment: Unlike other methods, clustering does not use the membrane normal direction, so the labels
upperandlowerleaflets are set somewhat arbitrarily following these rules:- First frame: The more populated cluster becomes the
upperleaflet. If equal, the cluster containing the lowest-index head identifier isupper. You can change this behavior using theflipkeyword. - Subsequent frames: Clusters are matched to previous frame's leaflets based on similarity.
- The matching is heuristic in membranes with lipid flip-flop and may fail if more than roughly 20% of lipids change leaflets between two consecutive analyzed frames. An error is raised if 20-80% lipids change leaflets. If more than 80% change leaflets, results will be incorrect without warning (though this is extremely unlikely).
- The matching may also fail if the spectral clustering identifies the leaflets incorrectly. In such case, you should provide the leaflet assignment manually.
- First frame: The more populated cluster becomes the
- Head identifier selection: When using the clustering method, always select head identifiers for all lipids in your membrane—even if analyzing only a specific subset of lipids and particularly when this subset resides in just one membrane leaflet.
- Extremely slow: Spectral clustering can be extremely slow, especially when your membrane is large. If you know that there is no flip-flop in your system, it is highly recommended to set the classification
frequencyto!Oncewhen using this method (see below).
Flipping the assignment
As mentioned above, the clustering method may in some cases mislabel the leaflets. For example, the leaflet you consider to be the lower leaflet of the membrane might be labeled as upper if it happens to contain more lipids. To correct this, you can add the flip keyword to invert the leaflet assignment during the analysis.
leaflets: !Clustering
heads: "name P"
flip: true
With this option enabled, any lipid that would normally be assigned to the upper leaflet based on the rules described above will instead be classified as part of the lower leaflet. Conversely, lipids that should be assigned to the lower leaflet will be actually assigned to the upper leaflet.
The
flipoption can be used with any leaflet classification method, but it is typically not very useful for anything else than the clustering method.
Classification frequency
By default, gorder performs leaflet classification independently for each analyzed frame. This ensures accurate analysis in membranes where lipid exchange occurs between leaflets. However, you can modify this behavior using the frequency keyword to specify how often leaflet classification should be performed.
Once
If you know that lipid flip-flop does not occur in your system and want to accelerate the analysis, you can use frequency: !Once. This option assigns lipids to individual membrane leaflets based on the first trajectory frame (not the TPR file structure), and this assignment is then used for all subsequent trajectory frames.
Example usage:
leaflets: !Local
membrane: "@membrane"
heads: "name P"
radius: 2.5
frequency: !Once
Using
frequency: !Onceis especially useful for the local and clustering classification methods which are computationally expensive.
Every N frames
Alternatively, you can specify that classification should occur every N analyzed trajectory frames using frequency: !Every N. For example, frequency: !Every 10 means that classification will be performed every 10 analyzed trajectory frames, with the closest previous assignment used for intermediate frames.
Example usage:
leaflets: !Global
membrane: "@membrane"
heads: "name P"
frequency: !Every 10
Important note: The frequency applies to analyzed trajectory frames. For instance, if the classification frequency is set to 10 and the analysis step size is 5 (see Analyzing a part of the trajectory), leaflet classification will occur every 50th (10×5) frame in the input trajectory.
Membrane normal
All leaflet classification methods (except for the clustering method) use the specified membrane normal to determine what is 'up' and what is 'down'. If your membrane is planar and aligned with the xy plane, no further action is needed. Otherwise, refer to this section of the manual.
Here, we just mention that the membrane normal used for leaflet classification can be decoupled from the 'global' membrane normal used for calculating order parameters:
leaflets: !Global
membrane: "@membrane"
heads: "name P"
membrane_normal: x # used only for leaflet classification
Leaflet-wise output
When a leaflet classification method is defined, gorder calculates order parameters for both the entire membrane and individual leaflets. Leaflet-specific order parameters are included in all gorder output formats: YAML, CSV, "table", and XVG.
During analysis, gorder also prints information about membrane composition in the first trajectory frame, allowing you to quickly check for any obvious errors. Such membrane composition information may look like this:
(...)
[*] Upper leaflet in the first analyzed frame: POPE: 45, POPC: 50, POPG: 5
[*] Lower leaflet in the first analyzed frame: POPE: 45, POPC: 50, POPG: 5
(...)
Below is an excerpt from an output YAML file containing results for individual membrane leaflets:
# Order parameters calculated with 'gorder v1.2.0' using a structure file 'system.tpr' and a trajectory file 'md.xtc'.
average order:
total: 0.1631
upper: 0.1629
lower: 0.1632
POPE:
average order:
total: 0.1601
upper: 0.1603
lower: 0.1598
order parameters:
POPE C22 (23):
total: 0.1036
upper: 0.1069
lower: 0.1003
bonds:
POPE H2R (24):
total: 0.0876
upper: 0.0924
lower: 0.0828
POPE H2S (25):
total: 0.1196
upper: 0.1214
lower: 0.1178
POPE C32 (32):
total: 0.2297
upper: 0.2291
lower: 0.2303
bonds:
POPE H2X (33):
total: 0.2423
upper: 0.241
lower: 0.2437
POPE H2Y (34):
total: 0.2171
upper: 0.2173
lower: 0.2168
#(...)
POPC:
average order:
total: 0.166
upper: 0.1654
lower: 0.1665
order parameters:
POPC C22 (32):
total: 0.1109
upper: 0.1117
lower: 0.1101
bonds:
POPC H2R (33):
total: 0.0935
upper: 0.0966
lower: 0.0904
POPC H2S (34):
total: 0.1283
upper: 0.1268
lower: 0.1297
POPC C32 (41):
total: 0.2373
upper: 0.236
lower: 0.2387
bonds:
POPC H2X (42):
total: 0.2483
upper: 0.2446
lower: 0.2519
POPC H2Y (43):
total: 0.2264
upper: 0.2273
lower: 0.2255
#(...)
POPG:
average order:
total: 0.1608
upper: 0.1621
lower: 0.1594
order parameters:
POPG C22 (25):
total: 0.0987
upper: 0.103
lower: 0.0944
bonds:
POPG H2R (26):
total: 0.08
upper: 0.0841
lower: 0.0759
POPG H2S (27):
total: 0.1174
upper: 0.1219
lower: 0.1129
POPG C32 (34):
total: 0.2272
upper: 0.2293
lower: 0.2251
bonds:
POPG H2X (35):
total: 0.2367
upper: 0.2391
lower: 0.2342
POPG H2Y (36):
total: 0.2177
upper: 0.2195
lower: 0.2159
#(...)
Maps of order parameters
Basic analysis
gorder can create maps of order parameters ("ordermaps") for the individual bond types and atom types. To construct an ordermap, the membrane plane is mapped onto a grid with bins of a specified size. Order parameters calculated for individual bonds are projected into the respective bins based on the position of the bond center. The average order parameter is then computed for each bin in the grid.
To enable the calculation of ordermaps, add the following section to your configuration YAML file:
ordermaps:
output_directory: "ordermaps"
gorder will generate one ordermap per bond type and save it in the ordermaps directory. If you calculate atomistic order parameters, ordermaps for individual heavy atom types will also be created as averages of the order parameters of heavy atom - hydrogen bonds. An ordermap with the average order parameter collected from all relevant bonds will also be written for each molecule type as well as for the entire system. If the calculation of order parameters for the individual membrane leaflets is requested (see Order parameters for individual leaflets), additional ordermaps for the upper and lower leaflets will be constructed.
If the output directory (here ordermaps) does not exist, it will be created. If it already exists, it will be backed up unless the overwrite option is enabled.
By default:
- The ordermap is placed in the plane perpendicular to the membrane normal (usually the xy-plane since the membrane normal is typically along the z-axis).
- The dimensions of the ordermaps are derived from the simulation box dimensions in the input structure file.
- The bin size is set to 0.1×0.1 nm.
- At least one sample per bin is required to calculate the order parameter for that bin.
You can manually override these default settings, see below.
Options for ordermaps
Plane of the ordermaps
To specify the plane for constructing the ordermaps, use the plane option:
ordermaps:
output_directory: "ordermaps"
plane: xz
The ordermaps will be created in the xz plane. For the xz plane, the first dimension corresponds to the x-axis and the second to the z-axis. For the yz plane, the first dimension corresponds to the z-axis and the second to the y-axis.
Dimensions of the ordermaps
To define the size (span) of the ordermaps, use the dim option:
ordermaps:
output_directory: "ordermaps"
dim:
- !Manual { start: 5.0, end: 10.0 }
- !Auto
In this example, the ordermaps are placed in the xy plane. They will cover an area spanning 5–10 nm along the x-axis, while the size along the y-axis will be automatically set based on the simulation box size.
Size of the bins
To set the bin size (granularity of the ordermaps), use the bin_size option:
ordermaps:
output_directory: "ordermaps"
bin_size: [0.05, 0.2]
Here, the bins are 0.05 nm wide along the x-axis and 0.2 nm wide along the y-axis.
Required number of samples
To specify the required number of samples per bin, use the min_samples option:
ordermaps:
output_directory: "ordermaps"
min_samples: 50
For this example, at least 50 samples must be collected in a bin to calculate the order parameter for it. If the collected sample count is less than 50, the order parameter for that bin will be recorded as NaN.
Ordermap format
Ordermaps are saved in a custom two-dimensional gorder format:
# Map of average order parameters calculated for the atom type POPC-C22-32.
# Calculated with 'gorder v1.2.0'.
@ xlabel x-dimension [nm]
@ ylabel y-dimension [nm]
@ zlabel order parameter ($-S_{CH}$)
@ zrange -1.0 0.5 0.2
$ type colorbar
$ colormap seismic_r
0.0000 0.0000 0.0663
0.0000 0.1000 0.0599
0.0000 0.2000 0.0890
0.0000 0.3000 0.0607
0.0000 0.4000 0.0851
0.0000 0.5000 0.0730
0.0000 0.6000 0.0635
0.0000 0.7000 0.0650
0.0000 0.8000 0.0823
0.0000 0.9000 0.1256
0.0000 1.0000 0.1154
(...)
0.1000 0.0000 0.0527
0.1000 0.1000 0.0738
0.1000 0.2000 0.0787
0.1000 0.3000 0.0735
0.1000 0.4000 0.0600
0.1000 0.5000 0.0745
0.1000 0.6000 0.0787
0.1000 0.7000 0.0897
0.1000 0.8000 0.1080
0.1000 0.9000 0.1279
0.1000 1.0000 0.1003
(...)
0.2000 0.0000 0.0645
0.2000 0.1000 0.1024
0.2000 0.2000 0.0880
0.2000 0.3000 0.0747
0.2000 0.4000 0.0647
0.2000 0.5000 0.0681
0.2000 0.6000 0.0839
0.2000 0.7000 0.1048
0.2000 0.8000 0.1208
0.2000 0.9000 0.1400
0.2000 1.0000 0.0974
(...)
This example ordermap corresponds to the atom type POPC-C22-32 (C22 of POPC). Lines beginning with # are comments, while lines starting with @ and $ define plotting parameters. Other lines contain the calculated order parameters. The first column represents the first coordinate of the bin (here x-dimension), the second column represents the second coordinate (here y-dimension), and the third column contains the average order parameter for the bin. The data is stored in row-major order. Note that the order parameter may be NaN; ensure your plotting software can handle such cases.
Plotting ordermaps
For easy visualization of ordermaps, gorder generates a Python script named plot.py in the ordermaps directory.
To run the script, it is recommended to use the uv package manager. To visualize an ordermap, navigate to the generated ordermaps directory and run:
uv run plot.py INPUT_ORDERMAP
Here, INPUT_ORDERMAP is the path to the ordermap file you want to plot. A window displaying the generated plot will appear.
If you want to plot and save the ordermap as a PNG file, run:
uv run plot.py INPUT_ORDERMAP OUTPUT_PNG
Here, OUTPUT_PNG is the path where the PNG file will be saved.
Examples of plotted ordermaps
When visualized, an ordermap for a specific atom type might look like this:
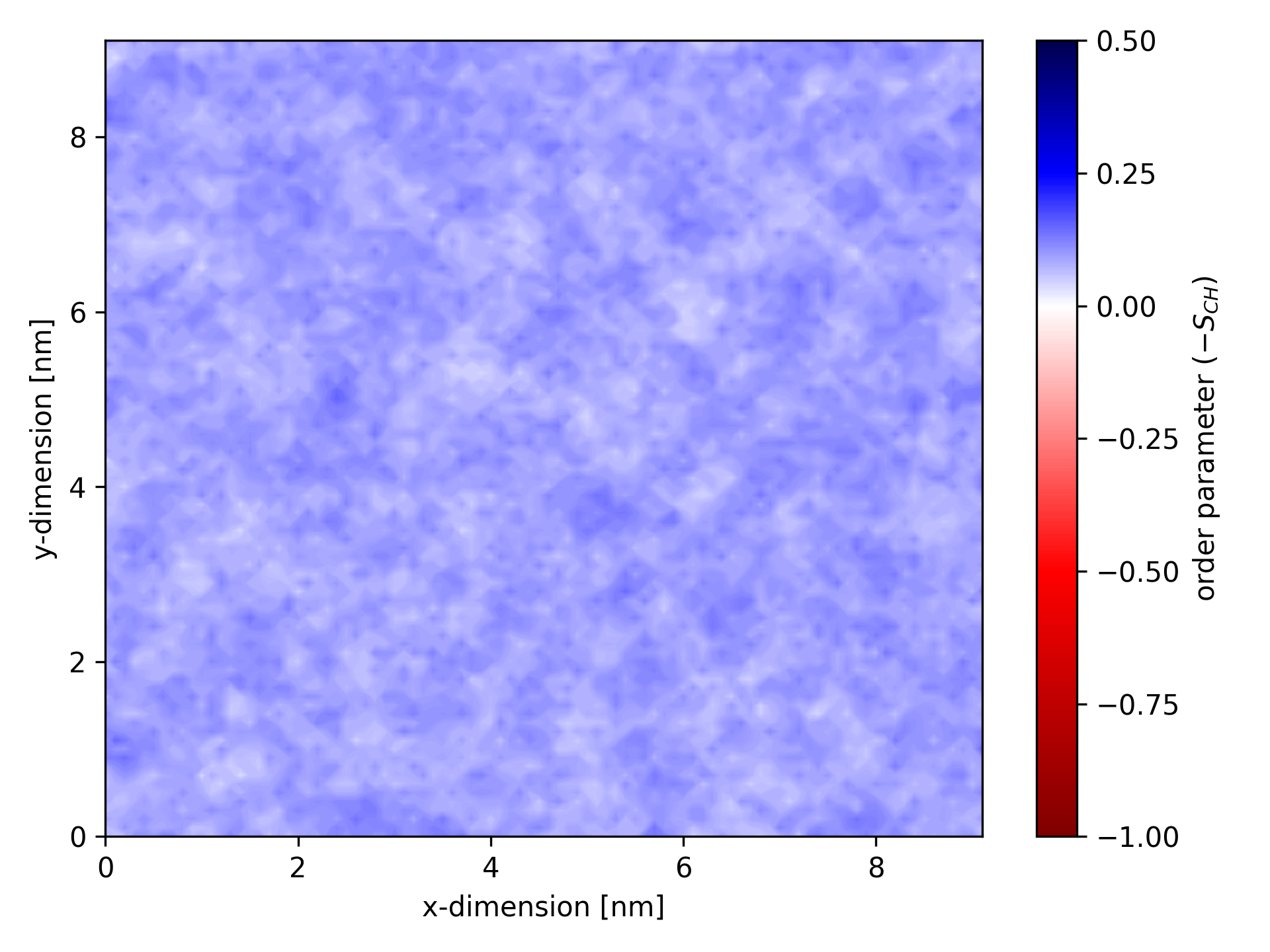
Ordermaps are especially useful for analyzing simulations involving transmembrane proteins. For such analyses, ensure the protein is centered and, ideally, RMSD-fitted in the trajectory.
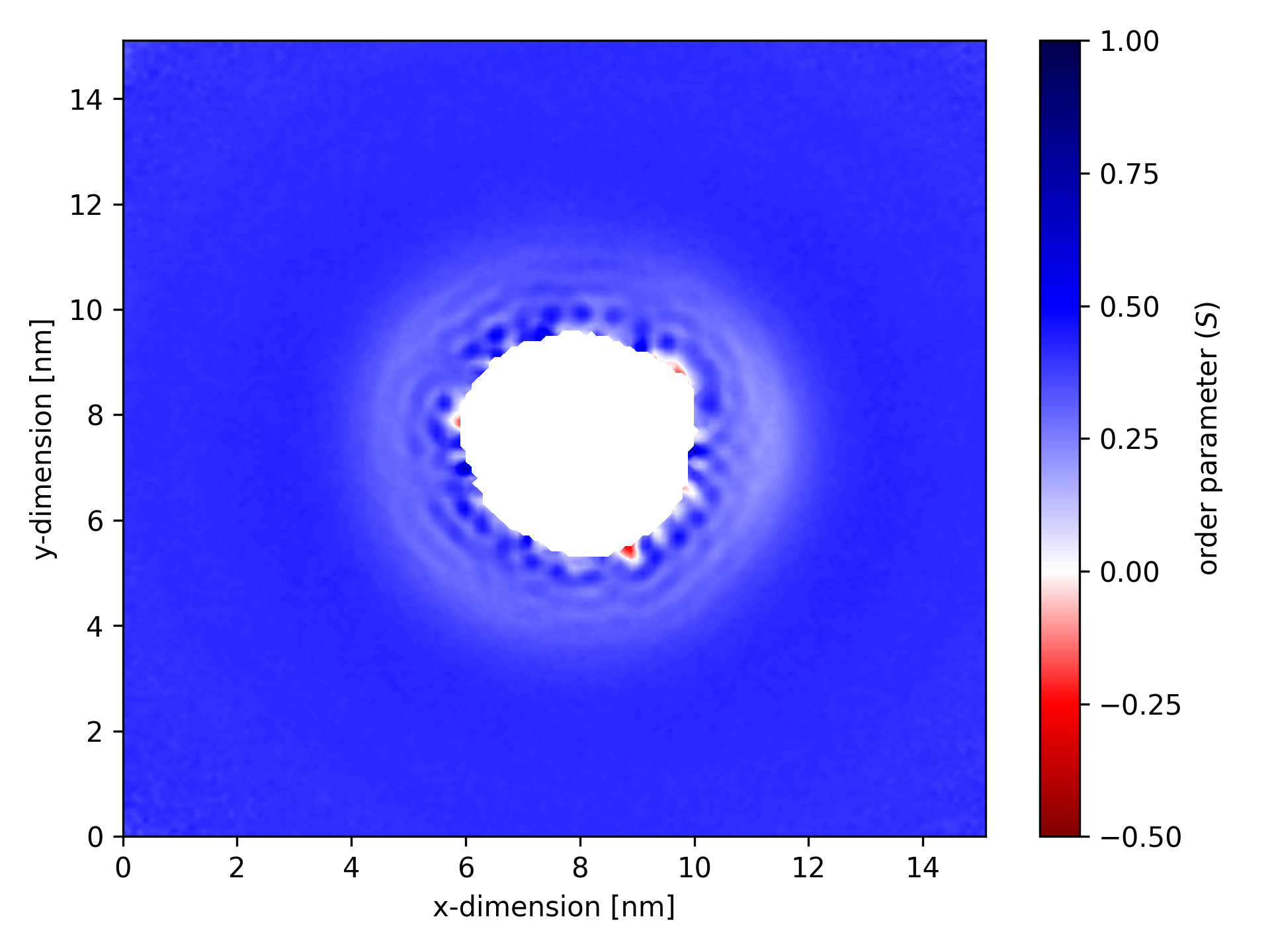
In this map of coarse-grained order for a particular lipid bond, the bright white region in the center represents the area occupied by the transmembrane protein. In this zone, no lipids (or an insufficient number to meet the min_samples threshold of 50) were detected. The membrane is disrupted in the vicinity of the protein.
Estimating errors
gorder is able to provide estimates of error for the individual bonds, atoms, and entire molecules.
The primary source of error when calculating order parameters from molecular dynamics simulations is the lack of convergence in the simulation. It’s also important to note that an order parameter calculated from a single trajectory frame is very unreliable unless the system is gigantic and contains many molecules. Using the standard deviation or standard error calculated from order parameters of individual frames or even samples is therefore not a viable option. Instead, gorder uses block averaging to calculate an error that reflects how well the simulation has converged.
To estimate the error, the trajectory is divided into blocks (default: 5). Each block represents a sequential portion of the trajectory: for example, the first fifth of the frames make up block #1, the second fifth forms block #2, and so on. The average order for each bond is calculated within each block, and a sample standard deviation is calculated from these block averages. This standard deviation is reported as the error. For simulations that are well-converged and have large enough blocks, the standard deviation between the blocks will be small.
Requesting error estimation
By default, gorder does not calculate error estimates. To enable error estimation, include the following line in your configuration YAML file:
estimate_error: true
The output YAML file will then include error estimates and may look similar to this:
# Order parameters calculated with 'gorder v1.2.0' using a structure file 'system.tpr' and a trajectory file 'md.xtc'.
average order:
total:
mean: 0.1631
error: 0.0014
POPE:
average order:
total:
mean: 0.1601
error: 0.0015
order parameters:
POPE C22 (23):
total:
mean: 0.1036
error: 0.0017
bonds:
POPE H2R (24):
total:
mean: 0.0876
error: 0.0054
POPE H2S (25):
total:
mean: 0.1196
error: 0.0033
POPE C32 (32):
total:
mean: 0.2297
error: 0.0045
bonds:
POPE H2X (33):
total:
mean: 0.2423
error: 0.0052
POPE H2Y (34):
total:
mean: 0.2171
error: 0.0045
#(...)
The errors are also reported in CSV and table files but not in XVG files:
molecule,residue,atom,relative index,total,total error,hydrogen #1,hydrogen #1 error,hydrogen #2,hydrogen #2 error,hydrogen #3,hydrogen #3 error
POPE,POPE,C22,23,0.1036,0.0017,0.0876,0.0054,0.1196,0.0033,,
POPE,POPE,C32,32,0.2297,0.0045,0.2423,0.0052,0.2171,0.0045,,
POPE,POPE,C23,35,0.2138,0.0034,0.2139,0.0025,0.2138,0.0047,,
POPE,POPE,C24,38,0.2165,0.0022,0.2158,0.0035,0.2172,0.0038,,
(...)
Molecule type POPE
TOTAL | HYDROGEN #1 | HYDROGEN #2 | HYDROGEN #3 |
C22 0.1036 ± 0.0017 | 0.0876 ± 0.0054 | 0.1196 ± 0.0033 | |
C32 0.2297 ± 0.0045 | 0.2423 ± 0.0052 | 0.2171 ± 0.0045 | |
C23 0.2138 ± 0.0034 | 0.2139 ± 0.0025 | 0.2138 ± 0.0047 | |
C24 0.2165 ± 0.0022 | 0.2158 ± 0.0035 | 0.2172 ± 0.0038 | |
(...)
Error estimates are currently not available for individual ordermaps.
Changing the number of blocks
If you are unsatisfied with the default number of blocks (5), you can adjust this parameter:
estimate_error:
n_blocks: 10
For example, setting n_blocks: 10 will use 10 blocks for error estimation instead of the default 5. You should avoid tweaking this number to get a lower error estimate.
Convergence analysis
gorder offers an additional method to assess simulation convergence by visualizing how the average order parameters evolve over time for individual molecules.
To request convergence analysis, include the following lines in your configuration YAML file:
estimate_error:
output_convergence: convergence.xvg
This will not only estimate errors as described above but also generate an XVG file (e.g., convergence.xvg) which shows the order parameters calculated for each frame as the average of values from that frame and all preceding frames (i.e., prefix averages).
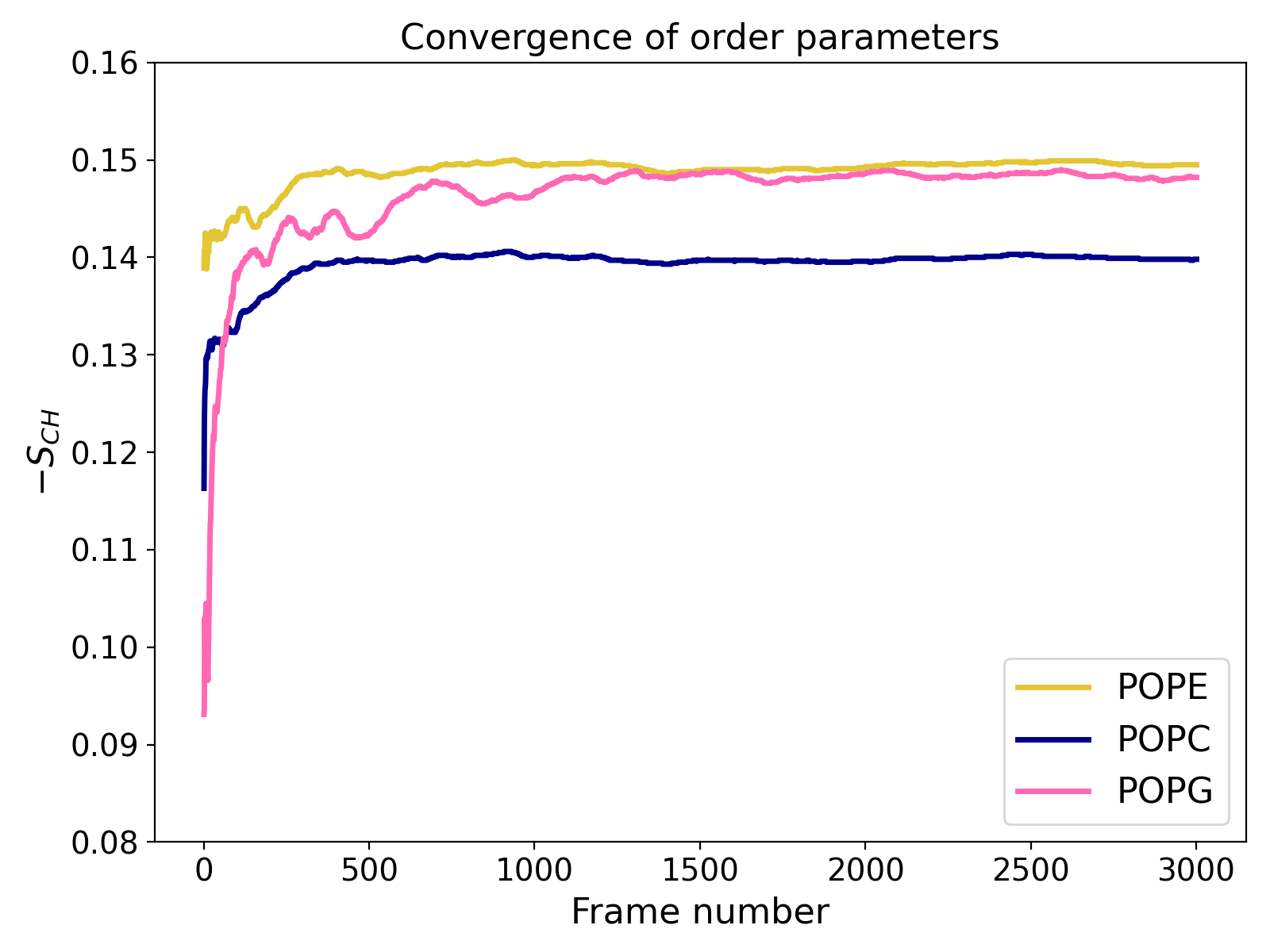
The example plot illustrates a relatively well-converged atomistic simulation where approximately 3,000 trajectory frames were analyzed. The chart shows that results for POPE and POPC lipids stabilize early (within the first third of the trajectory), while POPG converges more slowly (which is due to the smaller number of these lipids in the system).
Order parameters for a specific membrane region
gorder allows you to calculate order parameters for a specific region of the membrane within a defined geometric shape. Currently, three geometric shapes are supported for this selection: cuboid, cylinder, and sphere. When a geometric shape is specified, only bonds located within that shape are included in the order parameter calculations. The bond's inclusion is dynamically evaluated for every frame of the trajectory. This feature is useful for instance when analyzing order parameters near transmembrane proteins or specific membrane regions.
Note: The position of a bond is defined as the center of geometry of the bonded atoms.
Cuboidal selection
You can define a cuboid with specific dimensions and position to calculate order parameters for bonds within that region. To define a cuboid, include a configuration like the following in your configuration YAML file:
geometry: !Cuboid
x: [2.0, 4.0]
y: [1.0, 5.0]
z: [0.0, 3.0]
This configuration calculates order parameters for bonds where the x-coordinate is between 2 and 4 nm, the y-coordinate is between 1 and 5 nm, and the z-coordinate is between 0 and 3 nm.
If a dimension is not specified, the cuboid is considered unbounded in that dimension, allowing bonds at any coordinate along that axis:
geometry: !Cuboid
x: [2.0, 4.0]
y: [1.0, 5.0]
# the cuboid is infinite along the z-dimension
You can also define a reference point to make the cuboid dimensions relative instead of absolute:
geometry: !Cuboid
reference: [3.0, 1.0, 0.0]
x: [-2.0, 2.0]
y: [1.0, 5.0]
In this example, the cuboid extends from 1 to 5 nm along the x-dimension, from 2 to 6 nm along the y-dimension, and remains infinite along the z-dimension. Periodic boundary conditions are also taken into account when constructing the cuboid.
The reference point can be defined as a static coordinate, a dynamic center of geometry of a specified group of atoms, or the center of the simulation box. For details, see Specifying the reference point.
Below is an example of an ordermap where calculations considered only bonds within a specific cuboid. The static center of the cuboid is marked by a black ×.
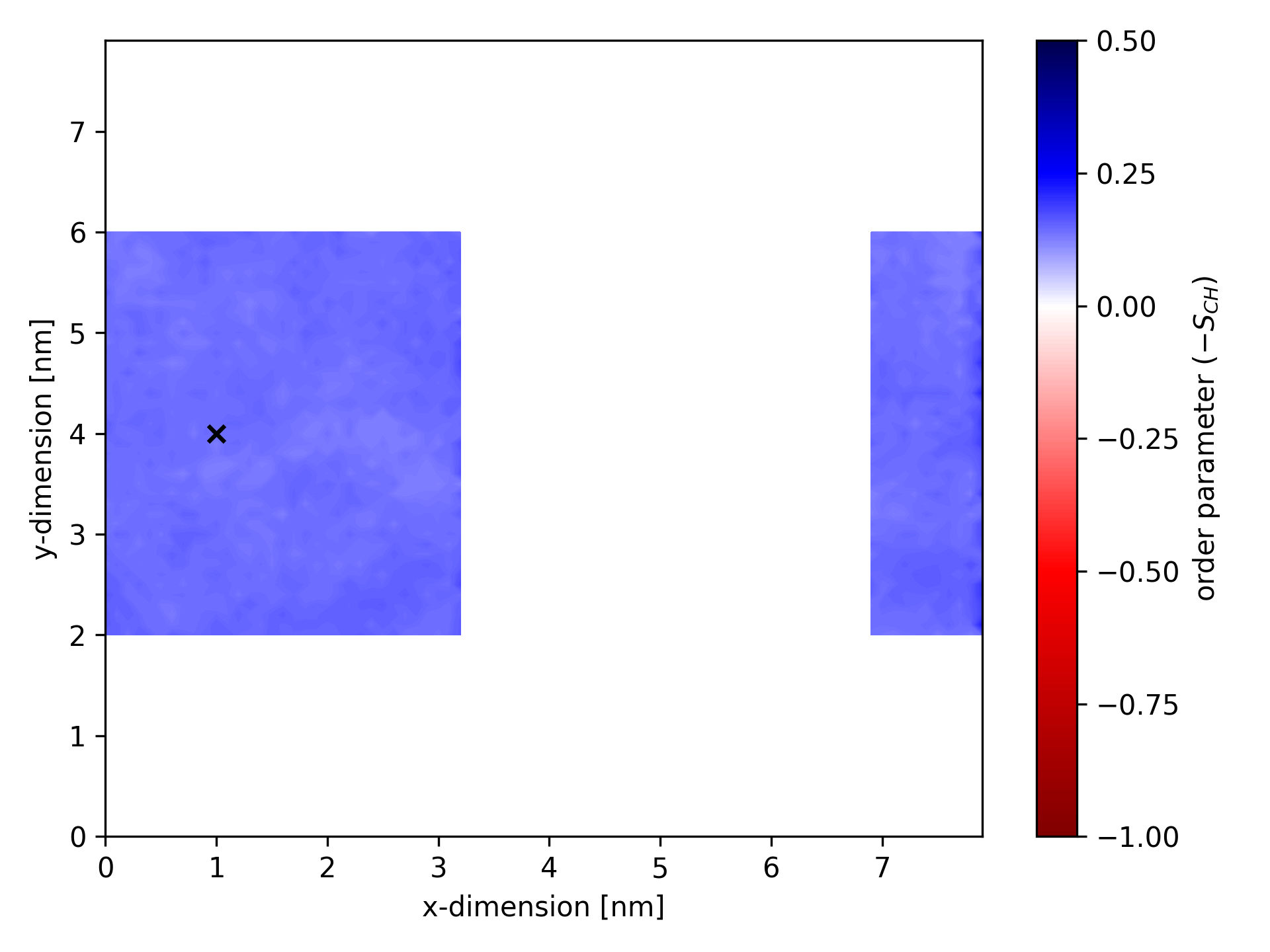
Cylindrical selection
You can define a cylinder with specific dimensions, orientation, and position to calculate order parameters for bonds within that region. To specify a cylinder, include a configuration like the following in your configuration YAML file:
geometry: !Cylinder
reference: [3.0, 1.0, 4.0]
radius: 2.5
span: [-1.0, 1.0]
orientation: z
This configuration calculates order parameters for bonds within a cylinder centered at x = 3 nm, y = 1 nm, z = 4 nm. The cylinder is oriented along the z-axis, has a radius of 2.5 nm, and a height of 2 nm (spanning from z = 3 nm to z = 5 nm). Periodic boundary conditions are also taken into account when constructing the cylinder.
The reference point for the cylinder can be defined as a static coordinate, a dynamic center of geometry of a specified group of atoms, or the center of the simulation box. If the reference value is omitted, the origin of the simulation box [0, 0, 0] is used. For more details, see Specifying the reference point.
If the span parameter is omitted, the cylinder is considered infinitely long along its main axis (orientation):
geometry: !Cylinder
radius: 2.5
orientation: z
In this case, the cylinder is centered at the origin [0, 0, 0], has a radius of 2.5 nm, is oriented along the z-axis, and has an infinite height.
The main axis of the cylinder (orientation) must align with one of the simulation box dimensions. Only x, y, and z orientations are supported.
Below is an example of an ordermap where calculations considered only bonds within a specific cylinder (oriented along the membrane normal). The static center of the cylinder is marked by a black ×.
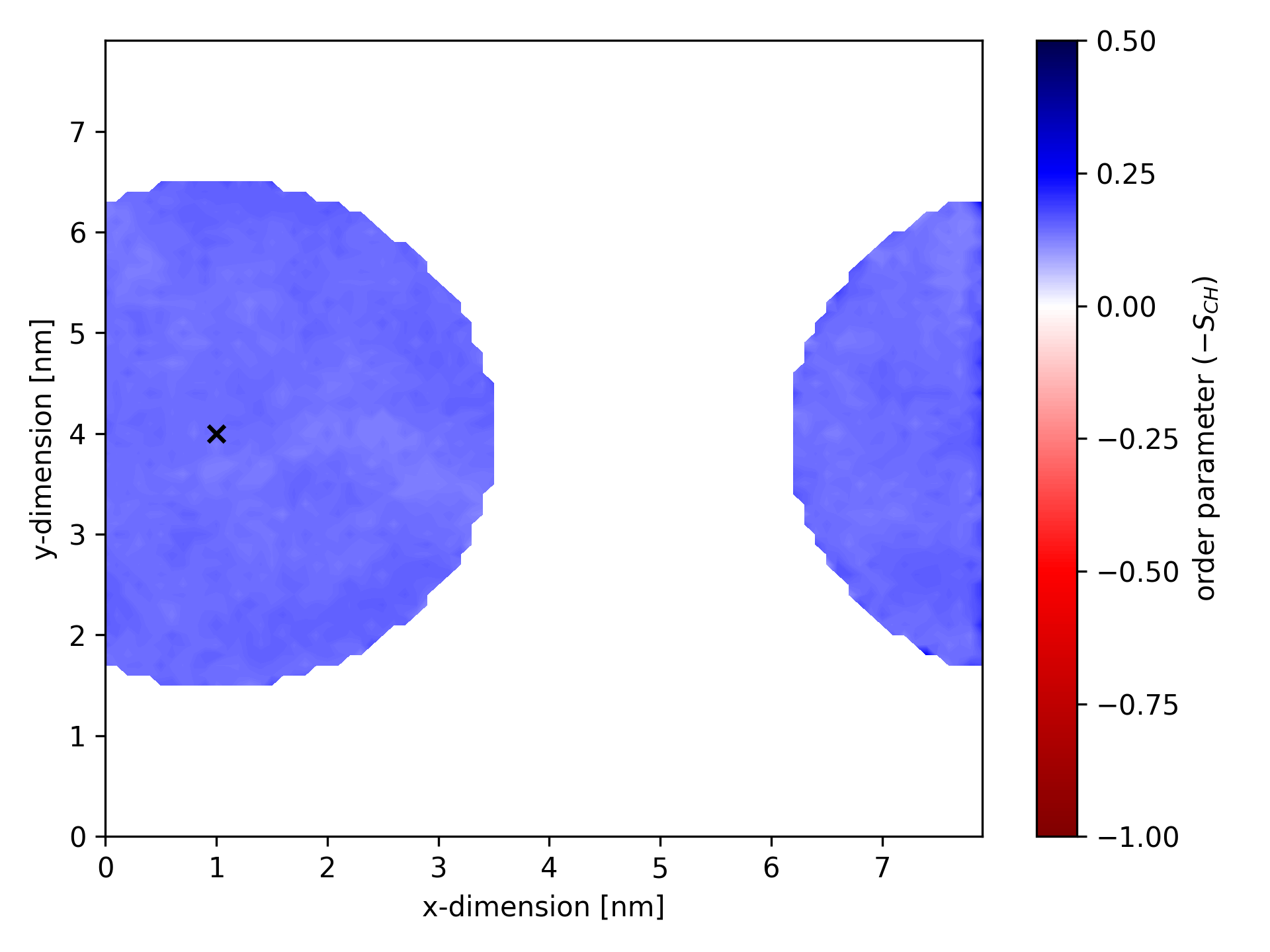
Spherical selection
You can define a sphere with a specific center and radius to calculate order parameters for bonds within that region. To specify a sphere, include a configuration like the following in your configuration YAML file:
geometry: !Sphere
reference: [3.0, 1.0, 4.0] # you can also use `center` instead of `reference`
radius: 2.5
This configuration calculates order parameters for bonds within a sphere centered at x = 3 nm, y = 1 nm, z = 4 nm, with a radius of 2.5 nm. Periodic boundary conditions are also considered when constructing the sphere.
The reference point (or center) for the sphere can be defined as a static coordinate, a dynamic center of geometry of a specified group of atoms, or the center of the simulation box. If the reference value is omitted, the sphere is centered at the origin of the simulation box [0, 0, 0]. For additional details, see Specifying the reference point.
Specifying the reference point
The reference point can be specified in one of three ways:
- Static coordinates
Define a fixed reference point using explicit coordinates:
reference: [2.0, 2.5, 1.5]
This places the reference point at x = 2.0 nm, y = 2.5 nm, and z = 1.5 nm. Note that these coordinates are not scaled by the simulation box dimensions.
- Dynamic center of geometry of selected atoms
Set the reference point as the center of geometry of a specified group of atoms:
reference: "@protein"
Here, the reference point corresponds to the geometric center of the @protein atom selection (all protein atoms in the system). The geometric center is recalculated for every frame of the trajectory. Use GSL syntax to specify the atom selection.
- Dynamic simulation box center
Choose the center of the simulation box as the reference point:
reference: !Center
The reference point will be the geometric center of the simulation box in all three dimensions, recalculated for every frame of the trajectory.
If no reference point is specified, the default is the simulation box origin at
[0, 0, 0].
Specifying membrane normal
gorder supports both static (provided by the user and applied to all lipid molecules throughout the entire analysis) and dynamic (calculated for each lipid molecule in each trajectory frame based on the actual membrane shape) membrane normal specification.
For planar membranes, the default static membrane normal is usually sufficient. However, for vesicles, you should always use dynamic membrane normal calculation. Keep in mind that computing the membrane normal dynamically is computationally expensive.
For complete control over membrane normals, you can also manually assign them for each molecule in every trajectory frame. For more details, refer to Manual membrane normals.
Static membrane normal
By default, gorder assumes the membrane normal is oriented along the z-axis, meaning the membrane is built in the xy-plane. If your membrane is oriented differently, you can manually specify the membrane normal in the configuration YAML file:
membrane_normal: x
In this example, the membrane normal is oriented along the x-axis, which means that the membrane is built in the yz-plane.
Only the primary axes of an orthogonal simulation box (
x,y, andz) are supported as static membrane normals.
Dynamic membrane normal
If your membrane is non-planar, or if you want to use the actual membrane normal instead of an idealized direction, you can enable dynamic membrane normal calculation. In this case, you must specify the selection of 'head identifier' atoms (similar to when assigning lipids to leaflets):
membrane_normal: !Dynamic
heads: "name P"
These 'head identifiers' are used to estimate the membrane surface and calculate the membrane normal for each lipid molecule. For consistency with leaflet classification, each analyzed lipid molecule must have exactly one head identifier; otherwise, an error will be raised.
The membrane normal is estimated for each lipid molecule in every frame. This is done by collecting the positions of 'head identifiers' located within a sphere around the molecule’s own head identifier and performing principal component analysis (PCA) on these positions. The last principal component (the direction with the least variation in atom positions) is then used as the membrane normal.
By default, the radius of the 'scanning sphere' is 2 nm, but you can adjust it in the input file:
membrane_normal: !Dynamic
heads: "name P"
radius: 1.5
The 'scanning sphere' must meet several requirements:
- It must be large enough to include a sufficient number of lipid heads for reasonable principal component analysis.
- It must be small enough to capture local membrane curvature correctly.
- Most importantly, it must be small enough to avoid including head identifiers from the opposite membrane leaflet, as this would distort the calculated membrane normal.
As a general rule of thumb, set the radius to approximately half of the membrane thickness.
Limitations
When using dynamic membrane normal calculation, you should be aware of some limitations:
-
Ignoring periodic boundary conditions
When ignoring periodic boundary conditions, membrane normals for lipids located near the edges (and especially at the corners) of the simulation box might not be calculated accurately. This occurs because, without PBC, not all nearby lipid heads are included in the surface estimation. If periodic boundary conditions are considered (which is the default), this issue does not occur. -
Assigning lipids to leaflets
All methods for classifying lipids into leaflets use the membrane normal to determine what is 'up' and what is 'down' in the membrane. However, due to technical limitations, these methods cannot use dynamically calculated membrane normals and always require a static membrane normal. You can specify this manually, for example:leaflets: !Global membrane: "@membrane" heads: "name P" membrane_normal: zThis membrane normal definition is used only when assigning lipids into leaflets. If you are working with a (reasonably) planar membrane, the membrane normal for leaflet classification does not need to be precise, so this approach is perfectly fine, even if you are otherwise working with dynamically calculated membrane normals. However, for a curved membrane, such as a vesicle, you should instead use the clustering method which does not use membrane normals or assign lipids into leaflets manually.
-
Constructing ordermaps
When constructing ordermaps, the plane in which an ordermap is generated is determined by the provided membrane normal. Since ordermaps can only be constructed in thexy,xz, oryzplane, they also require a static membrane normal (z,y, orx). If you are calculating the membrane normal dynamically and also want to construct ordermaps, you must specify the ordermap plane manually:ordermaps: output_directory: "ordermaps" plane: xy
Analyzing a part of the trajectory
By default, gorder analyzes the entire provided trajectory, meaning it processes all frames from the start to the end of the trajectory.
If you want to analyze only a specific part of the trajectory, you can use the begin, end, and step options to control the time range and frequency of analysis.
begin: Specifies the time (in ps) from which the trajectory will start being read.end: Specifies the time (in ps) at which the trajectory analysis will stop.step: Specifies the step size for reading trajectory frames (i.e., how often a frame will be analyzed).
Example: Calculating coarse-grained order parameters from a specific part of a trajectory
structure: system.tpr
trajectory: md.xtc
analysis_type: !CGOrder
beads: "@membrane"
begin: 100000.0
end: 300000.0
step: 5
output: order.yaml
In this example, the analysis starts at time 100,000 ps (100 ns) and ends at time 300,000 ps (300 ns). Every 5th frame of the trajectory will be analyzed.
Providing multiple trajectory files
If the trajectory you are analyzing is split across multiple files, you can provide all of them without having to manually merge them into a single file.
To do this, specify all your trajectory files as a list in your configuration YAML file:
structure: system.tpr
trajectory: [md1.xtc, md2.xtc, md3.xtc, md4.xtc, md5.xtc]
analysis_type: !AAOrder
heavy_atoms: "@membrane and name r'C3.+|C2.+'"
hydrogens: "@membrane and element name hydrogen"
output: order.yaml
gorder will seamlessly concatenate all the trajectory files and analyze them. If the individual trajectories are from the same simulation and follow each other directly, the last frame of a previous trajectory file will match the first frame of the subsequent file. Don't worry, gorder will automatically detect this scenario and consider only one of these frames. In contrast, if the frames have different simulation times, both will be included in the analysis. Note that if other frames in the trajectory share the same simulation time, gorder will analyze all of them. The tool only filters out duplicate frames at the boundaries between individual files.
Trajectory concatenation is supported only for XTC and TRR files. All files must be of the same trajectory type; concatenating XTC files with TRR files is not supported.
Alternatively, instead of listing all trajectory files, you can specify them using a glob pattern:
structure: system.tpr
trajectory: md*.xtc
analysis_type: !AAOrder
heavy_atoms: "@membrane and name r'C3.+|C2.+'"
hydrogens: "@membrane and element name hydrogen"
output: order.yaml
In this case, gorder will read all XTC files in the current directory whose names start with "md", such as md1.xtc, md2.xtc, md3.xtc, as well as mdXYZ.xtc, md239474.xtc, and similar files, if they exist.
Glob returns files in lexicographic order based on filenames. As a result,
md10.xtcmay appear beforemd2.xtc. Always verify the order of XTC files and ensure that filenames are structured so that lexicographic and numerical ordering align. For example, when dealing with trajectories 1–99, use filenames likemd01.xtctomd99.xtcto maintain the correct order.
Multithreaded analysis
The trajectory analysis can be performed using multiple threads. Utilizing more threads is generally much faster than using just one, although the speed gain also depends on the read performance of your storage device.
By default, gorder uses only 1 thread to perform the analysis. If you want to speed up the process by using multiple threads, you can specify the number of threads in your configuration YAML file:
n_threads: 4
In the example above, the analysis will be performed using 4 threads.
gorderguarantees binary-identical results on the same system and build environment, independent of the number of threads used.
Other options
Min samples
By default, gorder calculates order parameters for all bonds for which it collects at least one sample. If you want to increase the number of required samples, you can specify the minimum number of samples manually in the configuration YAML file:
min_samples: 10000
In this example, at least 10,000 samples are required for the bond to have a valid order parameter. If the number of samples is lower than this value, the order parameter will be NaN.
Overwrite
By default, gorder does not overwrite output files. Before writing an output file (or creating an output directory for the ordermaps), any existing file (or directory) with the same name is backed up. You can change this behavior by specifying the overwrite option in the configuration YAML file:
overwrite: true
In this example, no backups will be created, and files will be overwritten directly.
You can also specify this option on the command line when calling gorder:
$ gorder INPUT_YAML --overwrite
Silent
By default, gorder writes a lot of information about the progress of the analysis to the terminal (standard output). If you prefer not to see any of this information, you can specify it in the configuration YAML file:
silent: true
In this example, gorder will not write anything to standard output. Output files will still be generated, and the analysis will proceed as normal. If an error occurs, information about the error will be written to standard error output.
You can also specify this option on the command line when calling gorder:
$ gorder INPUT_YAML --silent
Manual lipid assignment and export
This chapter describes two advanced features related to leaflet classification: manual assignment and export.
Manually assigning lipids to leaflets
If you prefer not to use the automatic leaflet classification (described in Order parameters for individual leaflets)—for example, due to your system's complex geometry or a lack of confidence in gorder's assignment—you can manually specify the leaflet occupied by each lipid molecule for each analyzed frame.
There are two ways to manually assign lipids to membrane leaflets: using NDX files or a "leaflet assignment file".
Exporting leaflet assignment data
If you are using the built-in automatic leaflet classification, you may want to access the leaflet assignment results—for example, to verify that the assignment is correct or to reuse the lipid assignment in additional analyses. Instructions for doing this can be found in this section of the manual.
Assigning lipids using NDX file(s)
Using NDX files for lipid assignment is arguably a simpler method that enables seamless integration of gorder with FATSLiM, which implements one of the better lipid classification methods available.
NDX file
To begin, create an NDX file containing at least two groups. One group should contain lipid atoms (preferably just their 'head identifiers') of the upper leaflet, while the other group should contain lipid atoms of the lower leaflet.
[ UpperLeaflet ]
1140 1274 1810 2078 2212 2480 2614 2748 2882 3150 3284 3418 3686 3954 4088
4356 4490 4758 5026 5294 5428 5964 6098 6232 6366 6634 6902 7438 7572 7706
7974 8376 8778 8912 9046 9180 9582 9716 9850 10118 10386 10520 10654 10788 11056
11458 11592 11726 11860 12128 12262 12530 12664 13066 13200 13334 13602 14004 14272 14540
14808 14942 15076 15344 15478 15612 16014 16416 16818 17086 17354 17488 17622 17756 17890
18024 18158 18426 18694 18828 18962 19498 20034 20436 21106 21240 21508 21642 21776 21910
[ LowerLeaflet ]
1006 1408 1542 1676 1944 2346 3016 3552 3820 4222 4624 4892 5160 5562 5696
5830 6500 6768 7036 7170 7304 7840 8108 8242 8510 8644 9314 9448 9984 10252
10922 11190 11324 11994 12396 12798 12932 13468 13736 13870 14138 14406 14674 15210 15746
15880 16148 16282 16550 16684 16952 17220 18292 18560 19096 19230 19364 19632 19766 19900
20168 20302 20570 20704 20838 20972 21374 22044 22178 22312 22580 23116 23384 23518 23652
Using the NDX file
Then, specify the classification method in your configuration file:
leaflets: !FromNdx
ndx: "leaflets.ndx"
heads: "name P"
upper_leaflet: "UpperLeaflet"
lower_leaflet: "LowerLeaflet"
frequency: !Once
The leaflet assignment will be read from the NDX file leaflets.ndx, which should contain two groups: UpperLeaflet and LowerLeaflet. These groups should include the atoms of the upper and lower leaflets, respectively. The heads field specifies the head identifiers for all analyzed lipid molecules. Note that each lipid molecule must have only one head identifier; otherwise, an error will be returned. Since we are using a single NDX file, we also need to specify frequency: !Once, ensuring that the leaflet assignment from the NDX file is applied to all analyzed trajectory frames.
The UpperLeaflet and LowerLeaflet groups may contain various atoms, but they must include all heads atoms for the lipids being analyzed. In other words, every analyzed lipid must be assigned to a leaflet. The NDX file itself can contain any number of additional groups, but for performance reasons, it is recommended to keep both the file and the specified groups as small as possible.
Scrambling-safe leaflet assignment
Instead of using a single NDX file, you can provide multiple NDX files, such as one file per trajectory frame. Your configuration file may then look like this:
leaflets: !FromNdx
# first frame second frame third frame fourth frame
ndx: ["leaflets0001.ndx", "leaflets0002.ndx", "leaflets0003.ndx", "leaflets0004.ndx", (...)]
heads: "name P"
upper_leaflet: "UpperLeaflet"
lower_leaflet: "LowerLeaflet"
With this setup, leaflet assignment will be performed for each frame using a different NDX file.
Specifying NDX files using glob pattern
Instead of manually listing many NDX file paths in the configuration file, you can use glob pattern matching:
leaflets: !FromNdx
ndx: "leaflets*.ndx"
heads: "name P"
upper_leaflet: "UpperLeaflet"
lower_leaflet: "LowerLeaflet"
This pattern matches all NDX files in the current directory whose names start with "leaflets", such as leaflets0001.ndx, leaflets0002.ndx, as well as leafletsABCD.ndx, leafletsX.ndx, and similar files, if they exist.
Glob returns files in lexicographic order based on filenames. As a result,
leaflets10.ndxmay appear beforeleaflets2.ndx. Always verify the order of NDX files and ensure that filenames are structured so that lexicographic and numerical ordering align. For example, when dealing with frames 1–9999, use filenames likeleaflets0001.ndxtoleaflets9999.ndxto maintain the correct order.
Assignments for every Nth frame
If you want to specify leaflet assignments for every Nth analyzed frame (e.g., every 10th frame), follow these steps:
- Set the appropriate frequency in the configuration file. For example:
leaflets: !FromNdx
ndx: ["leaflets0001.ndx", "leaflets0002.ndx", "leaflets0003.ndx", "leaflets0004.ndx", (...)]
heads: "name P"
upper_leaflet: "UpperLeaflet"
lower_leaflet: "LowerLeaflet"
frequency: !Every 10
- Ensure that the number of provided NDX files exactly matches the number of analyzed frames divided by N. For example, if the trajectory is 10,000 frames long, every 5th frame is analyzed, and leaflet assignment is performed for every 10th analyzed frame, you must provide 200 NDX files. If you provide less or more NDX files, you will get an error during the analysis.
Assigning lipids using a leaflet assignment file
Another method for manually assigning lipids is using a YAML-formatted "leaflet assignment file". Unlike the NDX file method, where groups of atoms are used for assignment, the leaflet classification in this method is provided at the molecular level, with each molecule type having its own section in the file.
Leaflet assignment file
Consider a very small system consisting of 12 POPE molecules, 10 POPC molecules, and 8 POPG molecules. Let's say that this membrane is asymmetric:
- The upper leaflet contains 12 POPE molecules and 4 POPG molecules.
- The lower leaflet contains 10 POPC molecules and 4 POPG molecules.
The leaflet assignment file for this system should look as follows:
POPE:
- [Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper]
POPC:
- [Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower]
POPG:
- [Upper, Upper, Upper, Upper, Lower, Lower, Lower, Lower]
Each position in each list corresponds to the leaflet occupied by one lipid molecule of the specified type. The molecules are listed in the order in which their first atoms appear in the input structure file.
Note: You can substitute
UpperandLowerwith1and0, respectively, for specifying the 'upper' and 'lower' leaflets.
Using the leaflet assignment file
Save the leaflet assignment file, for example, as assignment.yaml. To use this file with gorder, include it in the YAML configuration file as follows:
leaflets: !FromFile
file: assignment.yaml
frequency: !Once
Ensure the frequency is set to
!Once, as this leaflet assignment file provides information for only a single frame.
Inline specification of leaflet assignment
You can also embed the leaflet assignment directly in the configuration file:
leaflets: !Inline
assignment:
POPE:
- [Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper]
POPC:
- [Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower]
POPG:
- [Upper, Upper, Upper, Upper, Lower, Lower, Lower, Lower]
frequency: !Once
Scrambling-safe leaflet assignment
If lipid flip-flop occurs in your system, you may want to specify the leaflet assignment for each analyzed frame. For example:
POPE:
# first analyzed frame
- [Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper]
# second analyzed frame
- [Upper, Upper, Lower, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper, Upper]
# third analyzed frame
- [Upper, Upper, Lower, Upper, Upper, Upper, Upper, Lower, Upper, Upper, Upper, Upper]
# (...)
POPC:
- [Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower]
- [Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Lower, Upper]
- [Lower, Lower, Lower, Upper, Lower, Lower, Lower, Lower, Lower, Upper]
# (...)
POPG:
- [Upper, Upper, Upper, Upper, Lower, Lower, Lower, Lower]
- [Upper, Upper, Upper, Upper, Lower, Lower, Lower, Lower]
- [Upper, Upper, Lower, Upper, Upper, Lower, Lower, Lower]
# (...)
To use this expanded file, specify it in the configuration YAML file:
leaflets: !FromFile
file: assignment.yaml
frequency: !Every 1
Alternatively, since frequency: !Every 1 is the default setting, you can simplify the configuration:
leaflets: !FromFile assignment.yaml
Assignments for every Nth frame
If you want to specify leaflet assignments for every Nth analyzed frame (e.g., every 10th frame), you must:
- Set the appropriate frequency in the configuration file. For example:
leaflets: !FromFile file: assignment.yaml frequency: !Every 10 - Ensure that the number of frames in the leaflet assignment file exactly matches the number of analyzed frames divided by N. For example, if the trajectory is 10,000 frames long, every 5th frame is analyzed, and leaflet assignment is performed for every 10th analyzed frame, the leaflet assignment file must contain information for 200 frames.
Note:
gorderis very opinionated when validating the leaflet assignment file. The file must specify the exact same number of lipid molecules for each molecule type as are present in the analyzed molecular system. Additionally, it must contain exactly the number of frames required for the leaflet assignment for the specified trajectory at the specified classification frequency (i.e., not a single unused frame can be present in the leaflet assignment file). The leaflet assignment file also cannot include information about molecule types that do not exist in the system. If any of these criteria are not met,gorderwill reject the leaflet assignment file. This strict validation reduces the chance of errors, as mistakes in manual assignments are easy to make but can be very difficult to detect otherwise.
Exporting lipid assignment data
You can configure gorder to export leaflet assignment data into an external file. This can be useful for verifying leaflet assignments, reusing them for additional analyses, or ensuring reproducibility.
To export the lipid assignment data, add the export keyword to the leaflets section of your configuration file and specify the path to the output file:
leaflets: !Global
membrane: "@membrane"
heads: "name P"
frequency: !Every 5
export: "leaflet_assignment.yaml" # or collect: "leaflet_assignment.yaml"
Alternatively, you can use the collect keyword instead of export.
Leaflet classification will be performed using the global method for every 5th analyzed frame (frequency: !Every 5). Once the trajectory has been processed, the assignment data will be written to leaflet_assignment.yaml. The data are exported in a condensed leaflet assignment file format. This means that you can directly reuse the exported YAML file in a subsequent gorder run by requesting manual leaflet classification. Since the output file is a valid YAML, you an also easily process it using other software.
Note that exporting lipid assignment data is only supported for automatic leaflet classification methods: global, local, individual, and clustering. Exporting lipid assignments is not supported for manual lipid assignment, as it is effectively pointless.
Below is an example of an exported leaflet assignment file for a very short trajectory:
# Leaflet assignment file generated by 'gorder v1.2.0' using a trajectory file 'md.xtc'.
POPE:
# Frame index 1
- [1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0]
# Frame index 6
- [1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0]
# Frame index 11
- [1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,0,0]
POPC:
# Frame index 1
- [1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0]
# Frame index 6
- [1,1,1,1,1,1,1,1,1,1,1,1,1,0,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0]
# Frame index 11
- [1,1,1,1,1,1,1,1,1,1,1,1,1,0,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0]
POPG:
# Frame index 1
- [0,0,0,0,0,1,1,1,1,1]
# Frame index 6
- [0,0,0,0,0,1,1,1,1,1]
# Frame index 11
- [0,0,0,0,0,1,1,1,1,1]
Data are written separately for each molecule type. Within each block, every line beginning with - corresponds to a single trajectory frame for which leaflet classification was performed.
Each number in the line represents the leaflet assignment of an individual lipid molecule of the specified type in the given trajectory frame:
1indicates the Upper leaflet0indicates the Lower leaflet
Molecules are listed in the order in which their first atom appears in the input structure file.
Note that the frame index refers to the sequential index of a frame in the input trajectory file, with the first analyzed frame assigned index 1. If your analysis begins later in the trajectory (using the begin keyword), frame 1 will not correspond to the very first frame of the trajectory.
Manual membrane normals assignment and export
This chapter describes two advanced features related to membrane normal(s): manual assignment of normals and export of dynamically calculated normals.
Assigning membrane normals manually
For absolute control over membrane normals, gorder allows manual specification of membrane normals for individual molecules in individual trajectory frames.
To assign membrane normals, you must first prepare a YAML-formatted membrane normals file. The structure of this file is similar to the "leaflet assignment file", which can be used to manually assign lipids to leaflets.
Membrane normals file
Consider a small system consisting of 12 POPE molecules, 10 POPC molecules, and 8 POPG molecules. We are analyzing a very short trajectory of just three frames.
The membrane normals file for this system might look like the following:
POPE:
# First frame
- - [0.45298508, -0.46554238, 0.7603123] # Membrane normal for the first POPE molecule
- [0.2806614, -0.15393148, 0.9473829] # Membrane normal for the second POPE molecule
- [0.43698093, 0.8893582, 0.13449812] # Membrane normal for the third POPE molecule
- [0.9750763, 0.21927911, -0.03380459]
- [0.92026454, -0.104457006, 0.37709686]
- [0.02029758, -0.70020616, -0.71365213]
- [0.40990028, -0.20503402, -0.88878727]
- [0.0019937176, -0.2997502, 0.9540157]
- [0.8564996, 0.47032556, 0.21260813]
- [0.049487855, 0.96054417, 0.27368945]
- [0.94329506, -0.10884861, 0.3136024]
- [0.088020176, 0.5334338, -0.8412495]
# Second frame
- - [0.79301494, -0.24298465, 0.5586464]
- [0.40446714, -0.19909416, 0.8926186]
- [0.52881354, 0.8245486, 0.20118622]
- [-0.87407386, -0.47928867, -0.07922962]
- [0.9209915, 0.08520295, 0.38015157]
- [0.3882479, -0.5707272, -0.7235565]
- [0.7917376, -0.2956609, -0.534543]
- [0.1430331, -0.5194693, 0.842433]
- [0.94468683, 0.19440018, 0.26415047]
- [0.02312048, 0.87527585, 0.4830711]
- [0.9681385, -0.2009886, 0.14937]
- [0.19331127, 0.4733897, -0.8593794]
# Third frame
- - [-0.5097876, 0.10576716, -0.8537739]
- [0.5334542, -0.14081219, 0.8340255]
- [0.4445155, 0.89264977, 0.07471584]
- [0.8339677, 0.37858394, -0.4014625]
- [0.8302863, -0.022482118, 0.5568835]
- [-0.20055892, 0.6154054, 0.76226795]
- [0.75300694, 0.113635726, -0.6481262]
- [0.38372648, -0.041432776, 0.9225169]
- [0.9821218, 0.18594055, -0.02937515]
- [-0.33398506, -0.92359644, -0.18821158]
- [0.9435965, 0.33108303, -0.0031385422]
- [0.18465553, 0.64861244, -0.73837954]
POPC:
# First frame
- - [0.83283865, 0.16904251, -0.5270716] # Membrane normal for the first POPC molecule
- [0.05139073, -0.5703374, -0.8198014] # Membrane normal for the second POPC molecule
- [0.9312034, -0.23388125, -0.2795709]
- [0.7521123, -0.58389324, 0.3056073]
- [0.112122506, 0.26633972, 0.9573357]
- [0.4494933, -0.8491345, 0.2773562]
- [0.27921712, -0.5408516, -0.7934214]
- [0.024991032, 0.5270191, 0.84948593]
- [0.290388, 0.68315774, 0.6700525]
- [0.3266205, 0.56352586, 0.75878704]
# Second frame
- - [0.831447, 0.03588961, -0.55444366]
# ...and 9 more membrane normals for the remaining POPC molecules
# Third frame
- - [0.9958207, -0.07853093, -0.046626113]
# ...and 9 more membrane normals for the remaining POPC molecules
POPG:
# Similar structure to POPE and POPC, but only 8 membrane normals per frame
Each innermost vector in the file represents the membrane normal assigned to a lipid molecule of the specified type. The molecules are listed in the order in which their first atoms appear in the input structure file. Each list of membrane normal vectors corresponds to one trajectory frame.
Unlike manual lipid assignment, membrane normals must be provided for every individual trajectory frame.
gorderstrictly validates the input membrane normals file. The file must contain exactly the same number of frames as the number of analyzed frames in the trajectory. Additionally, it must include the correct number of lipid molecules for each lipid type, and all lipid types being analyzed (and no additional ones) must be specified. Failure to meet these requirements will result in an error.
Using the membrane normals file
Save the membrane normals file, for example, as normals.yaml. To use this file with gorder, reference it in the YAML configuration file as follows:
membrane_normal: !FromFile normals.yaml
Specifying membrane normals inline
You can also define membrane normals directly within the configuration YAML file:
membrane_normal: !Inline
POPE:
- - [0.45298508, -0.46554238, 0.7603123]
- [0.2806614, -0.15393148, 0.9473829]
- [0.43698093, 0.8893582, 0.13449812]
- [0.9750763, 0.21927911, -0.03380459]
- [0.92026454, -0.104457006, 0.37709686]
- [0.02029758, -0.70020616, -0.71365213]
- [0.40990028, -0.20503402, -0.88878727]
- [0.0019937176, -0.2997502, 0.9540157]
- [0.8564996, 0.47032556, 0.21260813]
- [0.049487855, 0.96054417, 0.27368945]
- [0.94329506, -0.10884861, 0.3136024]
- [0.088020176, 0.5334338, -0.8412495]
# (...)
POPC:
# (...)
POPG:
# (...)
Exporting membrane normals
You can configure gorder to export dynamically calculated membrane normals into an external file. This can be useful for verifying the correctness of the calculation, reusing results for further analyses, or ensuring reproducibility.
To export dynamically calculated membrane normals, add the export keyword to the membrane_normal section of your configuration file and specify the path to the output file:
membrane_normal: !Dynamic
heads: "name P"
export: "normals.yaml" # or collect: "normals.yaml"
Alternatively, you can use the collect keyword instead of export.
The membrane normal is estimated from the shape of the membrane for each lipid molecule in every analyzed frame (for details, see this section of the manual). The calculated membrane normals are collected and written to normals.yaml. The data are exported in YAML format and can be directly reused in subsequent gorder run using membrane_normal: !FromFile normals.yaml. Since the output file is a valid YAML, you an also easily process it using other software.
Note that exporting membrane normals is only supported for dynamically calculated normals. You cannot export static normals (aligned with the
x,y, orzaxis) or manually assigned normals, since that would be pointless.
Below is an example of exported membrane normals from a very short trajectory of only a few lipids (part of a larger system that is ignored here for brevity):
# Membrane normals file generated by 'gorder v1.2.0' using trajectory file 'md.xtc'.
POPC:
# Frame index 1
- [[ 0.001220,-0.097192,-0.995265],[ 0.056732, 0.043663,-0.997434],[ 0.027993,-0.050879,-0.998312],[ 0.104689,-0.052774,-0.993104],[ 0.019110,-0.122940, 0.992230]]
# Frame index 2
- [[ 0.027892, 0.110019, 0.993538],[ 0.084509, 0.029770,-0.995978],[-0.040769, 0.034053, 0.998588],[ 0.134922,-0.062836,-0.988862],[ 0.020835,-0.139281, 0.990034]]
# Frame index 3
- [[ 0.036983, 0.024326, 0.999020],[ 0.113137,-0.034967,-0.992964],[-0.104049, 0.023076, 0.994304],[ 0.105232,-0.030009,-0.993995],[ 0.022746, 0.074999,-0.996924]]
POPE:
# Frame index 1
- [[ 0.000173,-0.064731,-0.997903],[ 0.097800,-0.069111,-0.992804],[ 0.004671, 0.137453, 0.990497],[ 0.041146,-0.053463,-0.997722],[ 0.002555,-0.003166, 0.999992]]
# Frame index 2
- [[ 0.037803,-0.048073,-0.998128],[ 0.058845,-0.095387,-0.993700],[ 0.007233, 0.204367, 0.978868],[-0.067056, 0.023346, 0.997476],[ 0.057067,-0.034264, 0.997782]]
# Frame index 3
- [[ 0.081806,-0.077921,-0.993598],[ 0.084231,-0.018487,-0.996275],[ 0.052119, 0.126407, 0.990608],[-0.071433,-0.016294, 0.997312],[ 0.046818,-0.043789, 0.997943]]
Data are written separately for each molecule type. Within each block, every line beginning with - corresponds to one analyzed trajectory frame. Each inner [x, y, z] vector represents the membrane normal for a specific molecule in that frame. Molecules are listed in the order in which their first atom appears in the input structure file.
Note: The above data come from a planar membrane built in the
xyplane. Therefore, thexandycomponents of each normal vector are close to0, while thezcomponents are close to±1. Thezcomponent may be close to+1or close to−1because a normal vector can point in two valid directions—in the case of a planar membrane, either "up" or "down". The orientation chosen bygorderis undefined and should not be relied upon. While this does not affect the calculated order parameters, you should keep it in mind if you plan to reuse the normal vectors in applications where their direction is important.
Missing membrane normals
When calculating membrane normals in a dynamically selected region of a membrane, normals are computed only for lipids that are actually needed—that is, lipids with at least one bond located within the specified region in a given frame. As a result, some membrane normals may be missing in the exported file.
Below is an example of exported membrane normals from the same system, but with order parameters calculated only inside a z-axis–oriented cylinder at the center of the membrane:
# Membrane normals file generated by 'gorder v1.2.0' using trajectory file 'md.xtc'.
POPC:
# Frame index 1
- [[.nan ,.nan ,.nan ],[ 0.056732, 0.043663,-0.997434],[.nan ,.nan ,.nan ],[.nan ,.nan ,.nan ],[ 0.019110,-0.122940, 0.992230]]
# Frame index 2
- [[ 0.027892, 0.110019, 0.993538],[ 0.084509, 0.029770,-0.995978],[.nan ,.nan ,.nan ],[.nan ,.nan ,.nan ],[ 0.020835,-0.139281, 0.990034]]
# Frame index 3
- [[ 0.036983, 0.024326, 0.999020],[ 0.113137,-0.034967,-0.992964],[.nan ,.nan ,.nan ],[.nan ,.nan ,.nan ],[.nan ,.nan ,.nan ]]
POPE:
# Frame index 1
- [[ 0.000173,-0.064731,-0.997903],[.nan ,.nan ,.nan ],[.nan ,.nan ,.nan ],[ 0.041146,-0.053463,-0.997722],[ 0.002555,-0.003166, 0.999992]]
# Frame index 2
- [[ 0.037803,-0.048073,-0.998128],[.nan ,.nan ,.nan ],[ 0.007233, 0.204367, 0.978868],[-0.067056, 0.023346, 0.997476],[ 0.057067,-0.034264, 0.997782]]
# Frame index 3
- [[ 0.081806,-0.077921,-0.993598],[.nan ,.nan ,.nan ],[.nan ,.nan ,.nan ],[-0.071433,-0.016294, 0.997312],[ 0.046818,-0.043789, 0.997943]]
Missing membrane normals are represented as [nan, nan, nan]. This allows you to identify which specific lipids were included in the calculation of order parameters in each frame. However, if you want to obtain all membrane normals, you must compute order parameters for the full set of lipids.
Using other input file formats
gorder is primarily designed for analyzing Gromacs simulations. The most efficient and straightforward way to use it is therefore by providing it with a TPR file and an XTC file. However, gorder also supports several other file formats for both structure/topology and trajectory files.
Structure and topology file formats
In rare cases where gorder cannot read your input TPR file, or if no TPR file is available, you can provide the system structure and topology using alternative file formats. gorder supports GRO, PDB, and PQR files as structure files.
For example, the following configuration YAML file is valid if the provided PDB file includes a connectivity section (and has fewer than 100,000 atoms due to PDB format limitations):
structure: system.pdb
trajectory: md.xtc
analysis_type: !CGOrder
beads: "@membrane"
output: order.yaml
If the PDB file lacks a connectivity section or if you are using a GRO or PQR file as the input structure, you must also supply a "bonds" file:
structure: system.gro
bonds: system.bnd # the file extension does not matter here
trajectory: md.xtc
analysis_type: !CGOrder
beads: "@membrane"
output: order.yaml
Connectivity information will be read from system.bnd. Note that the connectivity data in the bonds file overrides even information in the provided PDB or TPR file.
gorderidentifies the file format based on the file extension:.tprfor TPR files,.grofor GRO files,.pdbfor PDB files, and.pqrfor PQR files. Ensure that the file is named correspondingly.
Specification of the bonds file
The bonds file format is similar to the PDB connectivity block but simpler and much more flexible. It also supports systems of any (reasonable) size.
Each line in the file contains numbers separated by whitespace. The first number specifies the "target atom" (serial number), and the following numbers specify the atoms bonded to it. Serial numbers correspond to Gromacs numbering, where the first atom is 1, the second is 2, and so on (no matter the "atom number" in the input GRO or PDB file).
1 2 4
In this example, atom 1 is bonded to atoms 2 and 4. Atoms 2 and 4 are not bonded to each other.
Each bond needs to be specified only once (with any order of atoms), but duplicating entries is allowed. For instance, the following bonds files are equivalent:
1 2 4
2 3
1 2 4
2 1 3
3 2
4 1
(The system contains three bonds: atoms 1 and 2, atoms 1 and 4, and atoms 2 and 3.)
Bonds can be listed in any order, and multiple lines can start with the same target atom. Any number of bonds can be specified on one line. Empty lines are ignored, and any amount of whitespace is allowed between the numbers. Comments can be added using #:
1 2 4 # atom 1 is bonded to atoms 2 and 4
2 3
Here’s an excerpt from an example bonds file:
# Example bonds file for an atomistic system
1 2 3 4 5
2 1
3 1
4 1
5 1 6 7 8
6 5
7 5
8 5 9 10 15
9 8
10 8
11 12 13 14 15
12 11
13 11
14 11 16
15 8 11
16 14 17 18 19
# (...)
Note about selecting elements
When using the element keyword in GSL atom selection queries, atoms are selected based on their associated element. Element information is natively available in TPR files but is missing in other supported formats. If a non-TPR file is used, gorder will attempt to guess the elements of atoms based on the atom and residue names.
If gorder detects potential issues with the guess, it will display a warning in the terminal. You can then evaluate whether the concerns are harmless, avoid using the element keyword, or provide a TPR file instead.
Trajectory file formats
gorder is highly optimized to read XTC files as quickly as possible. If you have an XTC file or can generate one, it is recommended to use it. However, gorder also supports TRR and GRO trajectory file formats. You can use any of these files instead of an XTC trajectory, and gorder will automatically recognize the format based on the file extension.
gorderidentifies the file format based on the file extension:.xtcfor XTC files,.trrfor TRR files and.grofor GRO files. Ensure that your trajectory file is named accordingly.
Note that if you want to specify the analysis time range using begin and end while providing a GRO trajectory, the file must contain simulation time and step information in the 'title' line, formatted as follows:
Some Arbitrarily Long Title (...) t= SIMULATION_TIME step= SIMULATION_STEP
For example:
System t= 100.00000 step= 5000
Ignoring periodic boundary conditions
gorder fully supports the handling of orthogonal (i.e., rectangular cuboid) periodic boundary conditions (PBC) applied in all three dimensions. If your simulation box is orthogonal and PBC are applied in all three dimensions, you can use a raw trajectory generated by Gromacs directly, without worrying about making molecules whole in the simulation box or dealing with other PBC-related issues. gorder will handle everything automatically.
However, if your simulation box is not orthogonal, gorder cannot properly treat the system and you will get an error. In such cases, you must instruct gorder to ignore periodic boundary conditions by adding the following line to your configuration YAML file:
handle_pbc: false
When this option is set to false, gorder will disregard periodic boundary conditions and the presence of any simulation box. Effectively, it will treat the atoms in your system as if they exist in an infinite, unbound environment. When ignoring PBC, it is crucial to ensure that all lipid molecules in your system are whole in every frame of the input trajectory. If your molecules are "broken" at the box boundaries, you may silently get completely incorrect results! When ignoring PBC, you will always get a warning reminding you to make molecules whole:
[W] Periodic boundary conditions ignored. Lipid molecules must be made whole!
With Gromacs, you can make molecules whole using the following command:
gmx trjconv -s TPR_FILE -f XTC_FILE -pbc mol -o OUTPUT_XTC_FILE
The resulting OUTPUT_XTC_FILE should then be used as the trajectory input for gorder.
You can of course choose to ignore PBC for any system, even if it has an orthogonal box.
Note: You do not necessarily have to set
handle_pbctofalsewhen working with an orthogonal box with PBC applied in fewer than three dimensions. However, such systems are untested, so proceed with caution. In case you are not sure the analysis is correct, try using a trajectory with whole molecules, sethandle_pbctofalse, and check whether the results change.
Limitations when ignoring PBC
Be aware of the following limitations when PBC are ignored:
-
Order parameters for individual leaflets
Ensure that your membrane is fully within the simulation box. For example, if your membrane lies in the xy-plane, ensure it does not extend beyond the limits of the simulation box in the z-dimension or appear fragmented across the periodic boundary. Similarly, if your membrane is a vesicle, it should be located entirely and unbroken within the simulation box. Failing to meet this requirement may result in entirely incorrect lipid leaflet assignments. To avoid issues, you may want to center the membrane in the simulation box before running the analysis. -
Ordermap dimensions
When constructing ordermaps, you cannot automatically derive their dimensions from the simulation box because it is ignored. You must specify the dimensions manually. Forgetting to do this will result in an error. See this section of the manual for instructions on setting dimensions. -
Reference points for geometry selections
When calculating order parameters for a specific region, you cannot use the dynamic center of the simulation box as the reference point becausegorderignores box information. If this constraint is violated, you will get an error. However, you can still use static coordinates or the dynamic center of geometry as the reference point. If using the dynamic center of geometry, ensure that your selected atoms for center calculation are made whole, i.e., are not broken at the simulation box boundary. -
Using structures and trajectories not generated by Gromacs
Whilegorder(and Gromacs) interpret [0, 0, 0] as the origin of the simulation box (i.e., one of its corners), some other software treats [0, 0, 0] as the center of the simulation box. For basic analyses, this difference should not cause any issues, even if periodic boundaries are ignored, but you should still be aware of it. If you want to be completely sure that your analysis is correct, translate the system by half of the box size in each dimension so that [0, 0, 0] aligns with the origin of the simulation box (rather than its center) before running the analysis. -
Less efficient implementation of some calculations
When ignoring PBC, some optimization algorithms cannot be used as they rely on the presence of an orthogonal simulation box. As a result, you can expect the local method for leaflet classification and dynamic membrane normals calculation to be much slower, especially for very large systems.
Extracting all analysis options
When gorder runs, it automatically sets many analysis options to their default values if they are not explicitly specified. To view the full set of parameters used in an analysis, including defaults, you can run the gorder program with the --export-config argument. For example:
$ gorder analyze.yaml --export-config analyze_out.yaml
The file analyze_out.yaml will contain all the input parameters for the analysis, including:
- parameters specified in the input configuration file,
- parameters provided on the command line, and
- default values filled in by
gorder.
This output file can also be used as a valid input configuration file for subsequent gorder runs.
Using gorder as a Python package
gorder can also be used as a Python package, allowing you to call it directly from your Python code.
Installing using uv
The recommended way to install gorder as a Python package is to use the uv package manager. To add gorder to your uv project, run:
$ uv add git+https://github.com/Ladme/gorder.git#subdirectory=pygorder
Installing using pip
If you do not use uv, you can install gorder simply using pip:
$ pip install git+https://github.com/Ladme/gorder.git#subdirectory=pygorder
Using gorder
Once you install the package, import it into your Python code:
import gorder
Once imported, you can access all the options and functionality described in this manual.
For instance, the following configuration YAML file:
structure: system.tpr
trajectory: md.xtc
analysis_type: !CGOrder
beads: "@membrane"
output: order.yaml
can also by written as the following Python code:
analysis = gorder.Analysis(
structure = "system.tpr",
trajectory = "md.xtc",
analysis_type = gorder.analysis_types.CGOrder("@membrane"),
output_yaml = "order.yaml",
)
You can then run the analysis like this:
results = analysis.run()
and either write the results into the output file(s):
results.write()
or access them programmatically.
See the Python API documentation on ladme.github.io/pygorder-docs for more information about using gorder as a Python package.
Using gorder as a Rust crate
gorder can also be used as a Rust crate, allowing you to call it directly from your Rust code.
To use gorder in your project, first add it as a dependency:
$ cargo add gorder
Next, include the crate into your Rust code:
#![allow(unused)] fn main() { use gorder::prelude::*; }
Once imported, you can access all the options and functionality described in this manual.
For instance, the following configuration YAML file:
structure: system.tpr
trajectory: md.xtc
analysis_type: !CGOrder
beads: "@membrane"
output: order.yaml
can also by written as the following Rust code:
#![allow(unused)] fn main() { let analysis = Analysis::builder() .structure("system.tpr") .trajectory("md.xtc") .analysis_type(AnalysisType::cgorder("@membrane")) .output("order.yaml") .build()?; }
You can then run the analysis like this:
#![allow(unused)] fn main() { let results = analysis.run()?; }
and either write the results into the output file(s):
#![allow(unused)] fn main() { results.write()?; }
or access them programmatically.
See the Rust API documentation on docs.rs for more information about using gorder as a Rust crate.
Limitations
While gorder aims to provide accurate and reliable results, there are some known limitations and scenarios where it may not be suitable. The current limitations are as follows:
-
gordercannot read TPR files generated with Gromacs versions older than 5.1.- Why is this the case? This limitation stems from the
minitprcrate used to parse TPR files. The TPR file format evolves constantly, and supporting all ancient Gromacs versions is simply not feasible. - What can you do if your TPR file is this old? In most cases, the solution is simple: convert the old TPR file using a new version of Gromacs, i.e., run
gmx convert-tpr -s old_file.tpr -o new_file.tpr. If converting a TPR file is not an option for you, you can instead provide a PDB file with a connectivity section or a GRO file along with a bonds file (see Using other input file formats). - Will this be addressed in the future? Older Gromacs versions will likely never be fully supported.
- Why is this the case? This limitation stems from the
-
gorderonly fully supports systems with orthogonal simulation boxes and periodic boundary conditions applied in all three dimensions.- Why is this the case? This limitation arises from the
groan_rscrate, which provides only partial support for non-orthogonal simulation boxes. Handling periodic boundary conditions in such boxes is complex and not fully implemented at present. - What can you do if your box is not orthogonal? You must instruct
gorderto ignore periodic boundary conditions. Then, provide a trajectory where the lipid molecules are whole, and perform the analysis as usual. - What can you do if your system is not periodic in some dimensions? In most cases, there will be no issue; however, such systems have not been tested, and artifacts may occur. You can try ignoring periodic boundary conditions while ensuring the lipid molecules are whole and verify if the results change.
- Will this be addressed in the future?
gorderis unlikely to ever fully support non-orthogonal simulation boxes, simply because they are not common in membrane simulations. However, better support for partially periodic systems might be added in the future.
- Why is this the case? This limitation arises from the
-
gorderdoes not fully support the analysis of systems with multiple membranes.- Why is this the case? Handling multiple membranes is complex, and such systems are relatively uncommon.
gorderis primarily designed for analyzing single-membrane systems. This does not mean you cannot use it for multi-membrane systems, but some features may not work as expected (see below). - What can you do if your system contains multiple membranes? If you are performing basic analysis with a static membrane normal and no leaflet assignment, no special action is required. However, be aware that order parameters from all membranes will be averaged into a single collective set of order parameters. If you are assigning leaflets, note that the only usable automatic classification method is the
individualmethod. If you are using dynamic membrane normal calculation, ensure that the membranes are sufficiently far apart: specifically, farther than theradiusvalue used in the membrane normal calculation. You can of course also select atoms from a single membrane for analysis, but make sure to consistently apply this selection across all relevant sections, particularly in dynamic membrane normal calculation. - Will this be addressed in the future? Will
gorderever support membrane-specific order parameter calculations? This is highly unlikely. Multi-membrane systems are rare, and membrane-wise analysis can be performed by running individual analyses with different atom selections. Sincegorderis fast (and even faster when the trajectory is already cached), performing multiple runs should not be a significant issue.
- Why is this the case? Handling multiple membranes is complex, and such systems are relatively uncommon.
-
gorderfails when analyzing XTC trajectories from simulations longer than 2,147,483,647 simulation steps. (Note that this corresponds to simulation length of roughly 4.3 μs at 2 fs time step.)- Why is this the case? This limitation stems from the
mollycrate used to parse XTC files (and partially from the design of XTC files themselves). The simulation step in an XTC file is a 4-byte signed integer whose maximum value is 2,147,483,647. If a higher step is reached,mollycrashes with an error instead of overflowing like many other libraries. Read more here and here. - What can you do if your trajectory is this long? Remove the step information from your trajectory. A recommended approach is to use the step2frame tool, which generates a trajectory where step numbers are replaced with frame numbers. Use the converted trajectory as the input trajectory for
gorder. - Will this be addressed in the future? Yes — either by fixing the issue in the
mollycrate or by allowing compilation ofgorderwithoutmolly(which would, however, makegorderslower).
- Why is this the case? This limitation stems from the
Feedback
If you encounter an issue with the program, such as incorrect or unexpected behavior or functionality, please open an issue on GitHub or send an email to ladmeb@gmail.com.
For feature requests, concerns about calculation methods or metrics, or improvement suggestions, please use the same contact methods. If you discover software you believe outperforms gorder in lipid order calculations or simply does something better, sharing your feedback is encouraged. Any constructive criticism, even negative, is welcome.
If you enjoy using gorder, you can show your support by starring ★ the project on GitHub or recommending it to others.
Acknowledgements
gorder would not exist without the groundwork laid by many people across many projects. We extend our gratitude to the following:
-
The team behind the NMR Lipids project, whose deposited structures and trajectories were used in the validation of the order parameters and the testing of the analyses.
-
Patrick Fuchs and other developers of
buildH, whose incredibly detailed documentation has allowed the calculation of united-atom parameters to actually be reliable. -
Helgi Ingólfsson and Alex de Vries, who wrote the
do-orderscript, which served as the foundation for calculating coarse-grained order parameters. -
Sébastien Buchoux, the developer of the
FATSLiMpackage, which was an inspiration for dynamic membrane normal calculations. -
Marieke Westendorp, the developer of the
mollycrate, which is the main reasongorderis so fast. -
The developers of the Rust language and many of its crates, without which this project would be painful to complete.
-
The many people who contributed to the methodology of molecular dynamics simulations and their analysis over the years.
Citing
To cite the gorder software, please use the following reference:
Bartoš, L., Pajtinka, P., Vácha, R. (2025). gorder: Comprehensive tool for calculating lipid order parameters from molecular simulations. SoftwareX, 31, 102254. https://doi.org/10.1016/j.softx.2025.102254
BibTeX entry:
@article{gorder,
title = {gorder: Comprehensive tool for calculating lipid order parameters from molecular simulations},
journal = {SoftwareX},
volume = {31},
pages = {102254},
year = {2025},
issn = {2352-7110},
doi = {https://doi.org/10.1016/j.softx.2025.102254},
url = {https://www.sciencedirect.com/science/article/pii/S2352711025002213},
author = {Ladislav Bartoš and Peter Pajtinka and Robert Vácha},
}